The crystal structures of two ibuprofen sodium dihydrates, racemic sodium (
RS)-2-(4-isobutylphenyl)propanoate dihydrate or (
RS)-NaIBDH, Na
+·C
13H
17O
2-·2H
2O, and enantiomeric sodium (
S)-2-(4-isobutylphenyl)propanoate dihydrate or (
S)-NaIBDH, Na
+·C
13H
17O
2-·2H
2O, have been determined in the space groups
P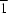
and
P1, respectively. The unit cells of the two triclinic structures have similar lattice parameters and cell volumes. The constituent ions have similar coordination environments, but differ slightly in their hydrogen-bonding interactions. The dominance of the interactions between the O atoms and the Na
+ cations explains the structural similarity of these two structures, despite the fact that one is heterochiral while the other is homochiral.
Supporting information
CCDC references: 285650; 285651
R/S-Ibuprofen sodium was purchased from Sigma (Saint Louis, Missouri). Enantiomeric S-ibuprofen sodium anhydrate was prepared by the method of Zhang et al. (2003). The respective dihydrate crystals of diffraction quality crystallized as thin plates by slow evaporation of a solution of the corresponding anhydrous sodium salt in water–acetonitrile (Ratio of solvents?). The crystals obtained were stored in contact with their mother liquor.
The absolute configuration was known for S-NaIBDH from the synthesis. This experiment did not attempt to determine the absolute structure. Friedel pairs were merged before refinement. The disordered iso-butyl group in (R/S)-NaIBDH was modeled as two fragments, while that in (S)-NaIBDH was modeled as one fragment, due to the instability of the constrained refinement as two fragments. The CH2–CH3 C—C distances in the iso-butyl group were constrained to be identical. The displacement parameters of all H atoms were constrained to be identical in each structure. Water H atoms were constrained to have identical O—H bond distances [Not identical in CIF tables?]. All other H atoms were refined using a riding model, with geometry idealized (C—H = 0.93–1.00 Å). [Please check added text.]
For both compounds, data collection: SMART (Bruker, 2001); cell refinement: SMART; data reduction: SAINT (Bruker, 2003); program(s) used to solve structure: SHELXS97 (Sheldrick, 1990); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: SHELXL97 and SHELXTL (Bruker, 2000); software used to prepare material for publication: PLATON (Spek, 2003).
(I) sodium (
RS)-
α-methyl [4-(isobutyl)phenyl]acetate dihydrate
top
Crystal data top
| Na+·C13H17O2−·2H2O | Z = 2 |
| Mr = 264.29 | F(000) = 284 |
| Triclinic, P1 | Dx = 1.202 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation, λ = 0.71073 Å |
| a = 5.7396 (4) Å | Cell parameters from 2507 reflections |
| b = 6.0284 (4) Å | θ = 1.8–27.0° |
| c = 23.8301 (17) Å | µ = 0.11 mm−1 |
| α = 83.457 (1)° | T = 173 K |
| β = 89.241 (1)° | Plate, colorless |
| γ = 63.154 (1)° | 0.40 × 0.30 × 0.10 mm |
| V = 730.20 (9) Å3 | |
Data collection top
Bruker SMART 1000 CCD area-detector
diffractometer | 3317 independent reflections |
| Radiation source: fine-focus sealed tube | 2610 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.037 |
| oscillation photo around ω at four ϕ scans | θmax = 27.6°, θmin = 1.7° |
Absorption correction: empirical (using intensity measurements)
based on ΔI (SADABS; Bruker, 2000) | h = −7→7 |
| Tmin = 0.791, Tmax = 1.0 | k = −7→7 |
| 8727 measured reflections | l = −30→30 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.048 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.135 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.0626P)2 + 0.4067P]
where P = (Fo2 + 2Fc2)/3 |
| 3317 reflections | (Δ/σ)max < 0.001 |
| 185 parameters | Δρmax = 0.49 e Å−3 |
| 12 restraints | Δρmin = −0.43 e Å−3 |
Crystal data top
| Na+·C13H17O2−·2H2O | γ = 63.154 (1)° |
| Mr = 264.29 | V = 730.20 (9) Å3 |
| Triclinic, P1 | Z = 2 |
| a = 5.7396 (4) Å | Mo Kα radiation |
| b = 6.0284 (4) Å | µ = 0.11 mm−1 |
| c = 23.8301 (17) Å | T = 173 K |
| α = 83.457 (1)° | 0.40 × 0.30 × 0.10 mm |
| β = 89.241 (1)° | |
Data collection top
Bruker SMART 1000 CCD area-detector
diffractometer | 3317 independent reflections |
Absorption correction: empirical (using intensity measurements)
based on ΔI (SADABS; Bruker, 2000) | 2610 reflections with I > 2σ(I) |
| Tmin = 0.791, Tmax = 1.0 | Rint = 0.037 |
| 8727 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.048 | 12 restraints |
| wR(F2) = 0.135 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.05 | Δρmax = 0.49 e Å−3 |
| 3317 reflections | Δρmin = −0.43 e Å−3 |
| 185 parameters | |
Special details top
Experimental. The single-crystal diffraction data were collected on a Bruker 1000 SMART CCD diffractometer at 173 (2) K. Oscillation photographs were taken around the ω axis at four different values of ϕ angle, 90 degrees apart. The data were indexed using SMART (Bruker, 2001) software and integrated with SAINT (Bruker, 2003) software. The structures were solved with direct methods and refined using the SHELXTL (Bruker, 2000) package. DSC employed 2920 MDSC (TA Instruments, New Castle, DE), while TGA employed 2950 T GA HR (TA Instruments, New Castle, DE), each at 5ϕC/min heating rate. |
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | Occ. (<1) |
| Na1 | 0.81780 (12) | 0.07127 (12) | 0.05758 (3) | 0.02261 (19) | |
| O4 | 0.3793 (2) | 0.2768 (2) | 0.08114 (5) | 0.0242 (3) | |
| O3 | −0.0100 (2) | 0.2966 (2) | 0.09916 (5) | 0.0241 (3) | |
| O2 | 0.7436 (2) | −0.2628 (2) | 0.03320 (5) | 0.0226 (3) | |
| O1 | 1.2485 (2) | −0.2238 (2) | 0.04190 (6) | 0.0257 (3) | |
| C1 | 0.2275 (3) | 0.2223 (3) | 0.11203 (7) | 0.0189 (3) | |
| C4 | 0.2462 (3) | 0.2303 (3) | 0.21566 (7) | 0.0236 (4) | |
| C2 | 0.3319 (3) | 0.0606 (3) | 0.16895 (7) | 0.0238 (4) | |
| C9 | 0.0595 (4) | 0.2266 (4) | 0.25309 (8) | 0.0284 (4) | |
| H9 | −0.0190 | 0.1202 | 0.2487 | 0.0487 (18)* | |
| C8 | −0.0136 (4) | 0.3757 (4) | 0.29668 (8) | 0.0326 (4) | |
| H8 | −0.1406 | 0.3687 | 0.3218 | 0.0487 (18)* | |
| C3 | 0.6273 (4) | −0.1048 (4) | 0.17190 (8) | 0.0331 (4) | |
| H3A | 0.6795 | −0.2115 | 0.2083 | 0.0487 (18)* | |
| H3B | 0.7167 | 0.0011 | 0.1682 | 0.0487 (18)* | |
| H3C | 0.6751 | −0.2100 | 0.1411 | 0.0487 (18)* | |
| C5 | 0.3531 (4) | 0.3925 (4) | 0.22260 (8) | 0.0286 (4) | |
| H5 | 0.4781 | 0.4014 | 0.1971 | 0.0487 (18)* | |
| C6 | 0.2792 (4) | 0.5412 (4) | 0.26629 (8) | 0.0316 (4) | |
| H6 | 0.3557 | 0.6495 | 0.2704 | 0.0487 (18)* | |
| C7 | 0.0949 (4) | 0.5347 (4) | 0.30430 (8) | 0.0316 (4) | |
| C10 | 0.0163 (5) | 0.6960 (4) | 0.35226 (9) | 0.0428 (5) | |
| H10A | −0.1025 | 0.6528 | 0.3764 | 0.0487 (18)* | 0.554 (5) |
| H10B | −0.0824 | 0.8737 | 0.3361 | 0.0487 (18)* | 0.554 (5) |
| H10D | 0.0783 | 0.8250 | 0.3445 | 0.0487 (18)* | 0.49 (5) |
| H10C | −0.1769 | 0.7842 | 0.3523 | 0.0487 (18)* | 0.38 (5) |
| H2E | 0.250 (5) | −0.043 (5) | 0.1729 (10) | 0.0487 (18)* | |
| H2B | 0.744 (5) | −0.279 (5) | −0.0010 (8) | 0.0487 (18)* | |
| H1A | 1.392 (4) | −0.226 (5) | 0.0444 (11) | 0.0487 (18)* | |
| H1B | 1.271 (5) | −0.367 (4) | 0.0535 (10) | 0.0487 (18)* | |
| H2A | 0.827 (5) | −0.401 (4) | 0.0525 (10) | 0.0487 (18)* | |
| C11 | 0.2456 (10) | 0.6679 (10) | 0.38922 (19) | 0.0571 (10) | 0.554 (5) |
| H11A | 0.3565 | 0.7221 | 0.3642 | 0.0487 (18)* | 0.554 (5) |
| C12 | 0.411 (2) | 0.4300 (16) | 0.4168 (4) | 0.0811 (13) | 0.554 (5) |
| H12A | 0.4815 | 0.3108 | 0.3890 | 0.0487 (18)* | 0.554 (5) |
| H12B | 0.3123 | 0.3757 | 0.4441 | 0.0487 (18)* | 0.554 (5) |
| H12C | 0.5554 | 0.4373 | 0.4367 | 0.0487 (18)* | 0.554 (5) |
| C13 | 0.1470 (14) | 0.8467 (13) | 0.4346 (2) | 0.0811 (13) | 0.554 (5) |
| H13A | 0.2959 | 0.8287 | 0.4577 | 0.0487 (18)* | 0.554 (5) |
| H13B | 0.0287 | 0.8059 | 0.4588 | 0.0487 (18)* | 0.554 (5) |
| H13C | 0.0534 | 1.0196 | 0.4164 | 0.0487 (18)* | 0.554 (5) |
| C11S | 0.1178 (13) | 0.5595 (12) | 0.4102 (2) | 0.0571 (10) | 0.45 |
| H11B | 0.0217 | 0.4567 | 0.4173 | 0.0487 (18)* | 0.446 (5) |
| C12S | 0.0164 (17) | 0.7407 (16) | 0.4547 (3) | 0.0811 (13) | 0.45 |
| H12D | 0.0845 | 0.6488 | 0.4923 | 0.0487 (18)* | 0.446 (5) |
| H12E | −0.1750 | 0.8189 | 0.4536 | 0.0487 (18)* | 0.446 (5) |
| H12F | 0.0747 | 0.8705 | 0.4469 | 0.0487 (18)* | 0.446 (5) |
| C13S | 0.391 (2) | 0.372 (2) | 0.4144 (5) | 0.0811 (13) | 0.45 |
| H13D | 0.4270 | 0.2696 | 0.3833 | 0.0487 (18)* | 0.446 (5) |
| H13E | 0.4271 | 0.2650 | 0.4506 | 0.0487 (18)* | 0.446 (5) |
| H13F | 0.5034 | 0.4562 | 0.4122 | 0.0487 (18)* | 0.446 (5) |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Na1 | 0.0164 (3) | 0.0218 (4) | 0.0299 (4) | −0.0080 (3) | 0.0013 (3) | −0.0067 (3) |
| O4 | 0.0197 (6) | 0.0223 (6) | 0.0271 (6) | −0.0068 (5) | 0.0027 (5) | −0.0017 (5) |
| O3 | 0.0192 (6) | 0.0238 (6) | 0.0294 (7) | −0.0096 (5) | −0.0032 (5) | −0.0032 (5) |
| O2 | 0.0201 (6) | 0.0215 (6) | 0.0258 (6) | −0.0088 (5) | −0.0013 (5) | −0.0038 (5) |
| O1 | 0.0151 (6) | 0.0206 (6) | 0.0404 (8) | −0.0076 (5) | 0.0004 (5) | −0.0016 (5) |
| C1 | 0.0203 (8) | 0.0145 (7) | 0.0215 (8) | −0.0066 (6) | 0.0002 (6) | −0.0066 (6) |
| C4 | 0.0230 (8) | 0.0234 (8) | 0.0197 (8) | −0.0068 (7) | −0.0024 (6) | −0.0006 (6) |
| C2 | 0.0248 (9) | 0.0222 (8) | 0.0227 (9) | −0.0094 (7) | −0.0019 (7) | −0.0017 (7) |
| C9 | 0.0275 (9) | 0.0298 (10) | 0.0267 (9) | −0.0121 (8) | −0.0005 (7) | −0.0024 (7) |
| C8 | 0.0289 (10) | 0.0379 (11) | 0.0264 (9) | −0.0112 (8) | 0.0059 (7) | −0.0039 (8) |
| C3 | 0.0279 (10) | 0.0301 (10) | 0.0270 (10) | −0.0007 (8) | −0.0044 (7) | −0.0020 (7) |
| C5 | 0.0292 (9) | 0.0309 (10) | 0.0265 (9) | −0.0141 (8) | 0.0035 (7) | −0.0044 (7) |
| C6 | 0.0359 (10) | 0.0303 (10) | 0.0307 (10) | −0.0162 (8) | −0.0014 (8) | −0.0058 (8) |
| C7 | 0.0342 (10) | 0.0288 (10) | 0.0229 (9) | −0.0062 (8) | −0.0010 (7) | −0.0050 (7) |
| C10 | 0.0523 (14) | 0.0382 (12) | 0.0324 (11) | −0.0137 (10) | 0.0055 (9) | −0.0131 (9) |
| C11 | 0.068 (3) | 0.063 (3) | 0.0354 (19) | −0.0216 (19) | 0.0001 (16) | −0.0210 (17) |
| C12 | 0.095 (2) | 0.084 (3) | 0.0519 (16) | −0.024 (2) | −0.0128 (14) | −0.0323 (17) |
| C13 | 0.095 (2) | 0.084 (3) | 0.0519 (16) | −0.024 (2) | −0.0128 (14) | −0.0323 (17) |
| C11S | 0.068 (3) | 0.063 (3) | 0.0354 (19) | −0.0216 (19) | 0.0001 (16) | −0.0210 (17) |
| C12S | 0.095 (2) | 0.084 (3) | 0.0519 (16) | −0.024 (2) | −0.0128 (14) | −0.0323 (17) |
| C13S | 0.095 (2) | 0.084 (3) | 0.0519 (16) | −0.024 (2) | −0.0128 (14) | −0.0323 (17) |
Geometric parameters (Å, º) top
| Na1—O3i | 2.3126 (13) | C5—H5 | 0.9500 |
| Na1—O4 | 2.3428 (13) | C6—C7 | 1.395 (3) |
| Na1—O1 | 2.3599 (14) | C6—H6 | 0.9500 |
| Na1—O2 | 2.3712 (14) | C7—C10 | 1.516 (3) |
| Na1—O1ii | 2.4127 (15) | C10—C11S | 1.500 (6) |
| Na1—Na1ii | 3.3773 (13) | C10—C11 | 1.527 (5) |
| O4—C1 | 1.263 (2) | C10—H10A | 0.9900 |
| O3—C1 | 1.258 (2) | C10—H10B | 0.9900 |
| O3—Na1iii | 2.3126 (13) | C10—H10D | 0.9900 |
| O2—H2B | 0.831 (17) | C10—H10C | 0.9900 |
| O2—H2A | 0.833 (17) | C11—C12 | 1.401 (10) |
| O1—Na1ii | 2.4127 (15) | C11—C13 | 1.533 (6) |
| O1—H1A | 0.819 (17) | C11—H11A | 1.0000 |
| O1—H1B | 0.828 (17) | C12—H12A | 0.9800 |
| C1—C2 | 1.530 (2) | C12—H12B | 0.9800 |
| C4—C9 | 1.391 (3) | C12—H12C | 0.9800 |
| C4—C5 | 1.393 (3) | C13—H13A | 0.9800 |
| C4—C2 | 1.522 (2) | C13—H13B | 0.9800 |
| C2—C3 | 1.528 (3) | C13—H13C | 0.9800 |
| C2—H2E | 0.93 (3) | C11S—C13S | 1.458 (12) |
| C9—C8 | 1.387 (3) | C11S—C12S | 1.527 (8) |
| C9—H9 | 0.9500 | C11S—H11B | 1.0000 |
| C8—C7 | 1.386 (3) | C12S—H12D | 0.9800 |
| C8—H8 | 0.9500 | C12S—H12E | 0.9800 |
| C3—H3A | 0.9800 | C12S—H12F | 0.9800 |
| C3—H3B | 0.9800 | C13S—H13D | 0.9800 |
| C3—H3C | 0.9800 | C13S—H13E | 0.9800 |
| C5—C6 | 1.387 (3) | C13S—H13F | 0.9800 |
| | | |
| O3i—Na1—O4 | 101.04 (5) | C11S—C10—C11 | 48.8 (3) |
| O3i—Na1—O1 | 88.45 (5) | C7—C10—C11 | 114.3 (2) |
| O4—Na1—O1 | 166.04 (5) | C11S—C10—H10A | 62.2 |
| O3i—Na1—O2 | 162.24 (5) | C7—C10—H10A | 108.7 |
| O4—Na1—O2 | 88.90 (5) | C11—C10—H10A | 108.7 |
| O1—Na1—O2 | 79.43 (5) | C11S—C10—H10B | 135.5 |
| O3i—Na1—O1ii | 106.65 (5) | C7—C10—H10B | 108.7 |
| O4—Na1—O1ii | 97.04 (5) | C11—C10—H10B | 108.7 |
| O1—Na1—O1ii | 89.92 (5) | H10A—C10—H10B | 107.6 |
| O2—Na1—O1ii | 86.43 (5) | C11S—C10—H10D | 108.4 |
| O3i—Na1—Na1ii | 100.70 (4) | C7—C10—H10D | 108.3 |
| O4—Na1—Na1ii | 139.96 (5) | C11—C10—H10D | 62.8 |
| O1—Na1—Na1ii | 45.59 (4) | H10A—C10—H10D | 141.9 |
| O2—Na1—Na1ii | 80.05 (4) | H10B—C10—H10D | 50.9 |
| O1ii—Na1—Na1ii | 44.33 (3) | C11S—C10—H10C | 108.4 |
| C1—O4—Na1 | 136.24 (11) | C7—C10—H10C | 108.4 |
| C1—O3—Na1iii | 125.73 (11) | C11—C10—H10C | 137.2 |
| Na1—O2—H2B | 116.8 (18) | H10A—C10—H10C | 51.4 |
| Na1—O2—H2A | 116.5 (18) | H10B—C10—H10C | 58.8 |
| H2B—O2—H2A | 111 (3) | H10D—C10—H10C | 107.4 |
| Na1—O1—Na1ii | 90.08 (5) | C12—C11—C10 | 118.3 (5) |
| Na1—O1—H1A | 134.1 (19) | C12—C11—C13 | 107.0 (5) |
| Na1ii—O1—H1A | 94.0 (18) | C10—C11—C13 | 110.2 (4) |
| Na1—O1—H1B | 110.1 (18) | C12—C11—H10D | 152.1 |
| Na1ii—O1—H1B | 121.4 (18) | C13—C11—H10D | 98.6 |
| H1A—O1—H1B | 106 (3) | C12—C11—H11A | 106.9 |
| O3—C1—O4 | 123.67 (15) | C10—C11—H11A | 106.9 |
| O3—C1—C2 | 116.90 (15) | C13—C11—H11A | 106.9 |
| O4—C1—C2 | 119.42 (14) | H10D—C11—H11A | 75.1 |
| C9—C4—C5 | 117.90 (17) | C11—C12—H12A | 109.5 |
| C9—C4—C2 | 120.84 (16) | C11—C12—H12B | 109.5 |
| C5—C4—C2 | 121.26 (16) | H12A—C12—H12B | 109.5 |
| C4—C2—C3 | 111.82 (15) | C11—C12—H12C | 109.5 |
| C4—C2—C1 | 108.66 (14) | H12A—C12—H12C | 109.5 |
| C3—C2—C1 | 113.59 (15) | H12B—C12—H12C | 109.5 |
| C4—C2—H2E | 108.8 (16) | C11—C13—H13A | 109.5 |
| C3—C2—H2E | 108.4 (16) | C11—C13—H13B | 109.5 |
| C1—C2—H2E | 105.3 (15) | H13A—C13—H13B | 109.5 |
| C8—C9—C4 | 121.01 (18) | C11—C13—H13C | 109.5 |
| C8—C9—H9 | 119.5 | H13A—C13—H13C | 109.5 |
| C4—C9—H9 | 119.5 | H13B—C13—H13C | 109.5 |
| C7—C8—C9 | 121.34 (18) | C13S—C11S—C10 | 115.6 (7) |
| C7—C8—H8 | 119.3 | C13S—C11S—C12S | 118.8 (6) |
| C9—C8—H8 | 119.3 | C10—C11S—C12S | 110.5 (5) |
| C2—C3—H3A | 109.5 | C13S—C11S—H11B | 103.2 |
| C2—C3—H3B | 109.5 | C10—C11S—H11B | 103.2 |
| H3A—C3—H3B | 109.5 | C12S—C11S—H11B | 103.2 |
| C2—C3—H3C | 109.5 | C11S—C12S—H12D | 109.5 |
| H3A—C3—H3C | 109.5 | C11S—C12S—H12E | 109.5 |
| H3B—C3—H3C | 109.5 | H12D—C12S—H12E | 109.5 |
| C6—C5—C4 | 120.82 (17) | C11S—C12S—H12F | 109.5 |
| C6—C5—H5 | 119.6 | H12D—C12S—H12F | 109.5 |
| C4—C5—H5 | 119.6 | H12E—C12S—H12F | 109.5 |
| C5—C6—C7 | 121.23 (18) | C11S—C13S—H13D | 109.5 |
| C5—C6—H6 | 119.4 | C11S—C13S—H13E | 109.5 |
| C7—C6—H6 | 119.4 | H13D—C13S—H13E | 109.5 |
| C8—C7—C6 | 117.68 (17) | C11S—C13S—H13F | 109.5 |
| C8—C7—C10 | 121.28 (19) | H13D—C13S—H13F | 109.5 |
| C6—C7—C10 | 121.05 (19) | H13E—C13S—H13F | 109.5 |
| C11S—C10—C7 | 115.6 (3) | | |
| Symmetry codes: (i) x+1, y, z; (ii) −x+2, −y, −z; (iii) x−1, y, z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H2A···O3iv | 0.83 (2) | 1.86 (2) | 2.6907 (17) | 176 (3) |
| O1—H1B···O4iv | 0.83 (2) | 1.98 (2) | 2.7998 (17) | 172 (3) |
| O1—H1A···O2i | 0.82 (2) | 1.95 (2) | 2.7507 (17) | 166 (3) |
| O2—H2B···O4v | 0.83 (2) | 2.04 (2) | 2.8438 (18) | 162 (3) |
| Symmetry codes: (i) x+1, y, z; (iv) x+1, y−1, z; (v) −x+1, −y, −z. |
(II) sodium S-
α-Methyl-4-(isobutyl)phenylacetate dihydrate
top
Crystal data top
| Na+·C13H17O2−·2H2O | Z = 2 |
| Mr = 264.29 | F(000) = 284 |
| Triclinic, P1 | Dx = 1.197 Mg m−3 |
| Hall symbol: P 1 | Mo Kα radiation, λ = 0.71073 Å |
| a = 5.7767 (5) Å | Cell parameters from 3725 reflections |
| b = 6.0835 (5) Å | θ = 1.9–27.0° |
| c = 23.880 (2) Å | µ = 0.11 mm−1 |
| α = 83.151 (1)° | T = 173 K |
| β = 86.482 (1)° | Plate, colorless |
| γ = 61.612 (1)° | 0.30 × 0.30 × 0.10 mm |
| V = 733.00 (11) Å3 | |
Data collection top
Bruker SMART 1000 CCD area-detector
diffractometer | 2589 independent reflections |
| Radiation source: fine-focus sealed tube | 2350 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.033 |
| oscillation photo around ω at four ϕ scans | θmax = 25.0°, θmin = 1.7° |
Absorption correction: empirical (using intensity measurements)
based on ΔI (SADABS; Bruker, 2000) | h = −6→6 |
| Tmin = 0.822, Tmax = 1.0 | k = −7→7 |
| 7383 measured reflections | l = −28→28 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.040 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.113 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.07 | w = 1/[σ2(Fo2) + (0.0737P)2 + 0.1763P]
where P = (Fo2 + 2Fc2)/3 |
| 2589 reflections | (Δ/σ)max < 0.001 |
| 356 parameters | Δρmax = 0.57 e Å−3 |
| 37 restraints | Δρmin = −0.30 e Å−3 |
Crystal data top
| Na+·C13H17O2−·2H2O | γ = 61.612 (1)° |
| Mr = 264.29 | V = 733.00 (11) Å3 |
| Triclinic, P1 | Z = 2 |
| a = 5.7767 (5) Å | Mo Kα radiation |
| b = 6.0835 (5) Å | µ = 0.11 mm−1 |
| c = 23.880 (2) Å | T = 173 K |
| α = 83.151 (1)° | 0.30 × 0.30 × 0.10 mm |
| β = 86.482 (1)° | |
Data collection top
Bruker SMART 1000 CCD area-detector
diffractometer | 2589 independent reflections |
Absorption correction: empirical (using intensity measurements)
based on ΔI (SADABS; Bruker, 2000) | 2350 reflections with I > 2σ(I) |
| Tmin = 0.822, Tmax = 1.0 | Rint = 0.033 |
| 7383 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.040 | 37 restraints |
| wR(F2) = 0.113 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.07 | Δρmax = 0.57 e Å−3 |
| 2589 reflections | Δρmin = −0.30 e Å−3 |
| 356 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Na1 | 0.8101 (2) | 1.0604 (2) | 0.74213 (6) | 0.0229 (3) | |
| O1' | 0.6838 (5) | 0.7400 (5) | 0.76335 (13) | 0.0271 (6) | |
| O3 | 0.4009 (5) | 1.3279 (5) | 0.70443 (12) | 0.0269 (6) | |
| O3' | 1.0901 (5) | 0.5648 (5) | 0.88818 (13) | 0.0325 (7) | |
| C1' | 1.2463 (7) | 0.6008 (7) | 0.91531 (16) | 0.0218 (8) | |
| C7' | 1.4239 (9) | 0.0957 (8) | 1.10835 (19) | 0.0349 (9) | |
| O4' | 1.4752 (5) | 0.5512 (5) | 0.89860 (12) | 0.0307 (6) | |
| O4 | 0.0412 (5) | 1.2862 (5) | 0.72135 (12) | 0.0278 (6) | |
| C4 | 0.1772 (8) | 1.3740 (7) | 0.58729 (16) | 0.0241 (8) | |
| C1 | 0.2351 (7) | 1.2636 (7) | 0.69105 (15) | 0.0196 (7) | |
| C2 | 0.2763 (7) | 1.1577 (7) | 0.63408 (16) | 0.0239 (8) | |
| H2E | 0.4662 | 1.0557 | 0.6285 | 0.065 (2)* | |
| C2' | 1.1606 (8) | 0.7045 (8) | 0.97265 (17) | 0.0270 (8) | |
| H2F | 1.2466 | 0.8063 | 0.9775 | 0.065 (2)* | |
| C4' | 1.2584 (7) | 0.4873 (7) | 1.01917 (16) | 0.0244 (8) | |
| C5' | 1.4482 (8) | 0.4513 (7) | 1.05721 (17) | 0.0306 (8) | |
| H5A | 1.5235 | 0.5577 | 1.0532 | 0.065 (2)* | |
| C8 | 0.2591 (8) | 1.6015 (8) | 0.50525 (17) | 0.0346 (9) | |
| H8 | 0.3792 | 1.6209 | 0.4801 | 0.065 (2)* | |
| C9' | 1.1546 (8) | 0.3191 (7) | 1.02605 (16) | 0.0297 (8) | |
| H9A | 1.0293 | 0.3358 | 1.0009 | 0.065 (2)* | |
| C9 | 0.3501 (8) | 1.4087 (7) | 0.54911 (16) | 0.0299 (8) | |
| H9 | 0.5299 | 1.3015 | 0.5528 | 0.065 (2)* | |
| C6 | −0.1804 (8) | 1.7322 (7) | 0.53724 (16) | 0.0319 (9) | |
| H6 | −0.3597 | 1.8416 | 0.5339 | 0.065 (2)* | |
| C6' | 1.5283 (8) | 0.2605 (8) | 1.10103 (17) | 0.0337 (9) | |
| H6A | 1.6547 | 0.2426 | 1.1261 | 0.065 (2)* | |
| C10 | −0.1055 (10) | 1.9738 (9) | 0.45030 (18) | 0.0469 (12) | |
| H10B | 0.0444 | 1.9669 | 0.4283 | 0.065 (2)* | |
| H10A | −0.1917 | 2.1339 | 0.4660 | 0.065 (2)* | |
| C5 | −0.0912 (7) | 1.5400 (7) | 0.58090 (16) | 0.0276 (8) | |
| H5 | −0.2113 | 1.5214 | 0.6062 | 0.065 (2)* | |
| C3 | 0.1498 (9) | 0.9882 (7) | 0.63103 (17) | 0.0347 (9) | |
| H3A | 0.1863 | 0.9241 | 0.5949 | 0.065 (2)* | |
| H3B | −0.0371 | 1.0834 | 0.6361 | 0.065 (2)* | |
| H3C | 0.2207 | 0.8508 | 0.6602 | 0.065 (2)* | |
| C3' | 0.8648 (8) | 0.8721 (8) | 0.97741 (18) | 0.0386 (10) | |
| H3D | 0.8229 | 0.9298 | 1.0142 | 0.065 (2)* | |
| H3E | 0.7750 | 0.7784 | 0.9717 | 0.065 (2)* | |
| H3F | 0.8102 | 1.0139 | 0.9493 | 0.065 (2)* | |
| C8' | 1.2363 (8) | 0.1287 (8) | 1.06984 (17) | 0.0329 (9) | |
| H18A | 1.1639 | 0.0199 | 1.0736 | 0.065 (2)* | |
| C7 | −0.0067 (9) | 1.7648 (8) | 0.49833 (17) | 0.0326 (9) | |
| C11 | −0.2987 (15) | 1.9620 (11) | 0.4107 (3) | 0.089 (2) | |
| H11 | −0.4547 | 1.9791 | 0.4325 | 0.065 (2)* | |
| C10' | 1.5115 (10) | −0.1107 (10) | 1.15591 (19) | 0.0470 (12) | |
| H10C | 1.6433 | −0.1022 | 1.1775 | 0.065 (2)* | |
| H10D | 1.5947 | −0.2708 | 1.1402 | 0.065 (2)* | |
| C13 | −0.383 (2) | 2.1818 (15) | 0.3643 (3) | 0.105 (3) | |
| H13A | −0.5117 | 2.1814 | 0.3407 | 0.065 (2)* | |
| H13B | −0.2328 | 2.1647 | 0.3420 | 0.065 (2)* | |
| H13C | −0.4582 | 2.3369 | 0.3812 | 0.065 (2)* | |
| C12 | −0.177 (2) | 1.7143 (14) | 0.3849 (3) | 0.127 (4) | |
| H12A | −0.1112 | 1.5778 | 0.4143 | 0.065 (2)* | |
| H12B | −0.0351 | 1.7032 | 0.3602 | 0.065 (2)* | |
| H12C | −0.3081 | 1.7055 | 0.3638 | 0.065 (2)* | |
| C11' | 1.2894 (13) | −0.1031 (11) | 1.1964 (2) | 0.088 (2) | |
| H11A | 1.1636 | −0.1220 | 1.1743 | 0.065 (2)* | |
| C12' | 1.141 (2) | 0.1438 (14) | 1.2220 (3) | 0.128 (4) | |
| H12D | 1.0486 | 0.2764 | 1.1928 | 0.065 (2)* | |
| H12E | 1.2628 | 0.1782 | 1.2399 | 0.065 (2)* | |
| H12F | 1.0170 | 0.1333 | 1.2494 | 0.065 (2)* | |
| O2 | 1.2223 (5) | 0.7038 (5) | 0.77081 (11) | 0.0231 (6) | |
| H2B | 1.204 (12) | 0.659 (12) | 0.8051 (10) | 0.065 (2)* | |
| C13' | 1.3995 (18) | −0.3255 (15) | 1.2423 (3) | 0.104 (3) | |
| H13D | 1.5092 | −0.3029 | 1.2672 | 0.065 (2)* | |
| H13E | 1.5014 | −0.4784 | 1.2251 | 0.065 (2)* | |
| H13F | 1.2567 | −0.3343 | 1.2634 | 0.065 (2)* | |
| O2' | 0.2274 (5) | 1.1389 (5) | 0.83852 (12) | 0.0246 (6) | |
| H2D | 0.197 (13) | 1.264 (8) | 0.856 (3) | 0.065 (2)* | |
| O1 | 0.7491 (5) | 1.1429 (5) | 0.83743 (12) | 0.0259 (6) | |
| Na1' | 0.6547 (2) | 0.8031 (2) | 0.86123 (6) | 0.0241 (3) | |
| H1A | 0.651 (11) | 1.266 (8) | 0.856 (2) | 0.065 (2)* | |
| H1C | 0.789 (10) | 0.611 (8) | 0.747 (3) | 0.065 (2)* | |
| H1D | 0.545 (7) | 0.725 (13) | 0.765 (3) | 0.065 (2)* | |
| H2C | 0.203 (12) | 1.198 (11) | 0.8041 (10) | 0.065 (2)* | |
| H2A | 1.277 (12) | 0.591 (9) | 0.748 (2) | 0.065 (2)* | |
| H1B | 0.904 (6) | 1.115 (12) | 0.844 (3) | 0.065 (2)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Na1 | 0.0199 (7) | 0.0215 (8) | 0.0306 (8) | −0.0126 (6) | 0.0003 (6) | −0.0021 (6) |
| O1' | 0.0206 (13) | 0.0238 (14) | 0.0409 (16) | −0.0128 (11) | 0.0028 (12) | −0.0092 (12) |
| O3 | 0.0209 (13) | 0.0266 (14) | 0.0354 (15) | −0.0115 (11) | −0.0043 (11) | −0.0074 (11) |
| O3' | 0.0250 (13) | 0.0281 (15) | 0.0415 (16) | −0.0075 (11) | −0.0061 (11) | −0.0132 (12) |
| C1' | 0.0211 (18) | 0.0152 (18) | 0.0253 (18) | −0.0060 (15) | −0.0009 (15) | 0.0002 (15) |
| C7' | 0.039 (2) | 0.029 (2) | 0.030 (2) | −0.0104 (18) | 0.0029 (18) | −0.0064 (17) |
| O4' | 0.0304 (15) | 0.0265 (14) | 0.0397 (16) | −0.0172 (12) | 0.0124 (12) | −0.0095 (12) |
| O4 | 0.0299 (14) | 0.0255 (14) | 0.0334 (15) | −0.0173 (12) | 0.0089 (12) | −0.0087 (11) |
| C4 | 0.031 (2) | 0.023 (2) | 0.0230 (19) | −0.0154 (17) | 0.0002 (15) | −0.0066 (16) |
| C1 | 0.0244 (18) | 0.0122 (16) | 0.0210 (17) | −0.0080 (14) | −0.0027 (15) | 0.0009 (14) |
| C2 | 0.0232 (18) | 0.0213 (19) | 0.0250 (19) | −0.0084 (15) | 0.0012 (15) | −0.0043 (15) |
| C2' | 0.033 (2) | 0.026 (2) | 0.0262 (19) | −0.0156 (17) | 0.0006 (16) | −0.0069 (16) |
| C4' | 0.0225 (18) | 0.024 (2) | 0.0249 (19) | −0.0089 (16) | 0.0039 (15) | −0.0069 (16) |
| C5' | 0.0299 (19) | 0.033 (2) | 0.032 (2) | −0.0161 (17) | 0.0010 (16) | −0.0078 (17) |
| C8 | 0.043 (2) | 0.040 (2) | 0.029 (2) | −0.027 (2) | 0.0078 (17) | −0.0048 (18) |
| C9' | 0.034 (2) | 0.034 (2) | 0.0249 (18) | −0.0178 (17) | −0.0018 (15) | −0.0049 (16) |
| C9 | 0.0301 (19) | 0.033 (2) | 0.030 (2) | −0.0178 (17) | 0.0005 (16) | −0.0055 (16) |
| C6 | 0.034 (2) | 0.031 (2) | 0.028 (2) | −0.0125 (17) | −0.0019 (16) | −0.0029 (16) |
| C6' | 0.031 (2) | 0.039 (2) | 0.028 (2) | −0.0128 (18) | −0.0045 (16) | −0.0049 (17) |
| C10 | 0.067 (3) | 0.035 (3) | 0.032 (2) | −0.021 (2) | 0.000 (2) | 0.004 (2) |
| C5 | 0.0271 (19) | 0.030 (2) | 0.0253 (18) | −0.0130 (16) | 0.0027 (15) | −0.0029 (15) |
| C3 | 0.054 (3) | 0.030 (2) | 0.029 (2) | −0.026 (2) | −0.0065 (18) | −0.0018 (16) |
| C3' | 0.039 (2) | 0.032 (2) | 0.030 (2) | −0.0053 (18) | 0.0066 (17) | −0.0037 (17) |
| C8' | 0.039 (2) | 0.031 (2) | 0.031 (2) | −0.0191 (18) | −0.0004 (16) | −0.0005 (16) |
| C7 | 0.045 (2) | 0.032 (2) | 0.0236 (19) | −0.020 (2) | −0.0030 (17) | −0.0012 (17) |
| C11 | 0.115 (6) | 0.088 (5) | 0.065 (4) | −0.054 (5) | −0.038 (4) | 0.035 (4) |
| C10' | 0.059 (3) | 0.038 (3) | 0.036 (2) | −0.017 (2) | −0.008 (2) | 0.004 (2) |
| C13 | 0.143 (8) | 0.093 (6) | 0.068 (5) | −0.051 (5) | −0.046 (5) | 0.035 (4) |
| C12 | 0.239 (13) | 0.103 (6) | 0.059 (4) | −0.095 (8) | −0.055 (6) | 0.009 (4) |
| C11' | 0.098 (5) | 0.092 (5) | 0.051 (4) | −0.034 (4) | 0.003 (3) | 0.022 (4) |
| C12' | 0.149 (8) | 0.102 (6) | 0.058 (4) | −0.008 (6) | 0.039 (5) | 0.014 (4) |
| O2 | 0.0217 (13) | 0.0215 (14) | 0.0281 (14) | −0.0118 (11) | 0.0004 (11) | −0.0023 (11) |
| C13' | 0.124 (7) | 0.097 (6) | 0.066 (4) | −0.041 (5) | 0.006 (4) | 0.032 (4) |
| O2' | 0.0219 (13) | 0.0216 (14) | 0.0307 (14) | −0.0099 (11) | −0.0018 (11) | −0.0048 (11) |
| O1 | 0.0215 (13) | 0.0253 (14) | 0.0358 (15) | −0.0143 (12) | 0.0026 (11) | −0.0067 (12) |
| Na1' | 0.0204 (7) | 0.0235 (8) | 0.0309 (8) | −0.0126 (6) | −0.0006 (6) | −0.0011 (6) |
Geometric parameters (Å, º) top
| Na1—O3 | 2.302 (3) | C10—C7 | 1.516 (6) |
| Na1—O4i | 2.327 (3) | C10—C11 | 1.539 (5) |
| Na1—O1 | 2.358 (3) | C10—H10B | 0.9700 |
| Na1—O1' | 2.379 (3) | C10—H10A | 0.9700 |
| Na1—O2 | 2.404 (3) | C5—H5 | 0.9300 |
| Na1—Na1' | 3.3623 (16) | C3—H3A | 0.9600 |
| O1'—Na1' | 2.396 (3) | C3—H3B | 0.9600 |
| O1'—H1C | 0.849 (17) | C3—H3C | 0.9600 |
| O1'—H1D | 0.849 (18) | C3'—H3D | 0.9600 |
| O3—C1 | 1.264 (4) | C3'—H3E | 0.9600 |
| O3'—C1' | 1.255 (5) | C3'—H3F | 0.9600 |
| O3'—Na1' | 2.313 (3) | C8'—H18A | 0.9300 |
| C1'—O4' | 1.259 (5) | C11—C12 | 1.518 (5) |
| C1'—C2' | 1.531 (5) | C11—C13 | 1.531 (5) |
| C7'—C6' | 1.386 (6) | C11—H11 | 0.9800 |
| C7'—C8' | 1.390 (6) | C10'—C11' | 1.544 (5) |
| C7'—C10' | 1.500 (6) | C10'—H10C | 0.9700 |
| O4'—Na1'i | 2.300 (3) | C10'—H10D | 0.9700 |
| O4—C1 | 1.255 (5) | C13—H13A | 0.9600 |
| O4—Na1ii | 2.327 (3) | C13—H13B | 0.9600 |
| C4—C9 | 1.384 (5) | C13—H13C | 0.9600 |
| C4—C5 | 1.397 (6) | C12—H12A | 0.9600 |
| C4—C2 | 1.523 (6) | C12—H12B | 0.9600 |
| C1—C2 | 1.531 (5) | C12—H12C | 0.9600 |
| C2—C3 | 1.531 (5) | C11'—C12' | 1.515 (5) |
| C2—H2E | 0.9800 | C11'—C13' | 1.533 (5) |
| C2'—C4' | 1.522 (6) | C11'—H11A | 0.9800 |
| C2'—C3' | 1.523 (6) | C12'—H12D | 0.9600 |
| C2'—H2F | 0.9800 | C12'—H12E | 0.9600 |
| C4'—C5' | 1.386 (6) | C12'—H12F | 0.9600 |
| C4'—C9' | 1.402 (6) | O2—H2B | 0.849 (17) |
| C5'—C6' | 1.384 (6) | O2—H2A | 0.849 (17) |
| C5'—H5A | 0.9300 | C13'—H13D | 0.9600 |
| C8—C7 | 1.383 (6) | C13'—H13E | 0.9600 |
| C8—C9 | 1.392 (6) | C13'—H13F | 0.9600 |
| C8—H8 | 0.9300 | O2'—Na1' | 2.380 (3) |
| C9'—C8' | 1.382 (6) | O2'—H2D | 0.849 (17) |
| C9'—H9A | 0.9300 | O2'—H2C | 0.849 (17) |
| C9—H9 | 0.9300 | O1—Na1' | 2.376 (3) |
| C6—C5 | 1.386 (5) | O1—H1A | 0.849 (17) |
| C6—C7 | 1.393 (6) | O1—H1B | 0.849 (18) |
| C6—H6 | 0.9300 | Na1'—O4'ii | 2.300 (3) |
| C6'—H6A | 0.9300 | | |
| | | |
| O3—Na1—O4i | 103.28 (11) | C2—C3—H3B | 109.5 |
| O3—Na1—O1 | 102.33 (11) | H3A—C3—H3B | 109.5 |
| O4i—Na1—O1 | 92.95 (10) | C2—C3—H3C | 109.5 |
| O3—Na1—O1' | 90.03 (11) | H3A—C3—H3C | 109.5 |
| O4i—Na1—O1' | 165.27 (11) | H3B—C3—H3C | 109.5 |
| O1—Na1—O1' | 90.27 (11) | C2'—C3'—H3D | 109.5 |
| O3—Na1—O2 | 165.58 (11) | C2'—C3'—H3E | 109.5 |
| O4i—Na1—O2 | 87.31 (10) | H3D—C3'—H3E | 109.5 |
| O1—Na1—O2 | 86.63 (10) | C2'—C3'—H3F | 109.5 |
| O1'—Na1—O2 | 78.53 (10) | H3D—C3'—H3F | 109.5 |
| O3—Na1—Na1' | 101.66 (8) | H3E—C3'—H3F | 109.5 |
| O4i—Na1—Na1' | 134.88 (8) | C9'—C8'—C7' | 121.6 (4) |
| O1—Na1—Na1' | 44.96 (7) | C9'—C8'—H18A | 119.2 |
| O1'—Na1—Na1' | 45.46 (8) | C7'—C8'—H18A | 119.2 |
| O2—Na1—Na1' | 76.60 (7) | C8—C7—C6 | 117.8 (4) |
| O3—Na1—H1B | 118.1 (11) | C8—C7—C10 | 121.1 (4) |
| O4i—Na1—H1B | 79.5 (11) | C6—C7—C10 | 121.1 (4) |
| O1—Na1—H1B | 18.8 (7) | C12—C11—C13 | 110.3 (6) |
| O1'—Na1—H1B | 100.0 (13) | C12—C11—C10 | 111.6 (6) |
| O2—Na1—H1B | 73.1 (12) | C13—C11—C10 | 109.0 (5) |
| Na1'—Na1—H1B | 55.6 (12) | C12—C11—H11 | 108.6 |
| Na1—O1'—Na1' | 89.50 (10) | C13—C11—H11 | 108.6 |
| Na1—O1'—H1C | 110 (5) | C10—C11—H11 | 108.6 |
| Na1'—O1'—H1C | 128 (4) | C7'—C10'—C11' | 114.7 (4) |
| Na1—O1'—H1D | 139 (4) | C7'—C10'—H10C | 108.6 |
| Na1'—O1'—H1D | 93 (4) | C11'—C10'—H10C | 108.6 |
| H1C—O1'—H1D | 101 (6) | C7'—C10'—H10D | 108.6 |
| C1—O3—Na1 | 125.6 (2) | C11'—C10'—H10D | 108.6 |
| C1'—O3'—Na1' | 134.9 (3) | H10C—C10'—H10D | 107.6 |
| O3'—C1'—O4' | 123.6 (4) | C11—C13—H13A | 109.5 |
| O3'—C1'—C2' | 119.2 (3) | C11—C13—H13B | 109.5 |
| O4'—C1'—C2' | 117.2 (3) | H13A—C13—H13B | 109.5 |
| C6'—C7'—C8' | 117.4 (4) | C11—C13—H13C | 109.5 |
| C6'—C7'—C10' | 121.1 (4) | H13A—C13—H13C | 109.5 |
| C8'—C7'—C10' | 121.5 (4) | H13B—C13—H13C | 109.5 |
| C1'—O4'—Na1'i | 132.1 (2) | C11—C12—H12A | 109.5 |
| C1—O4—Na1ii | 136.2 (2) | C11—C12—H12B | 109.5 |
| C9—C4—C5 | 118.1 (4) | H12A—C12—H12B | 109.5 |
| C9—C4—C2 | 121.0 (4) | C11—C12—H12C | 109.5 |
| C5—C4—C2 | 121.0 (3) | H12A—C12—H12C | 109.5 |
| O4—C1—O3 | 123.4 (3) | H12B—C12—H12C | 109.5 |
| O4—C1—C2 | 119.8 (3) | C12'—C11'—C13' | 111.1 (6) |
| O3—C1—C2 | 116.8 (3) | C12'—C11'—C10' | 112.7 (6) |
| C4—C2—C3 | 111.5 (3) | C13'—C11'—C10' | 110.3 (5) |
| C4—C2—C1 | 109.1 (3) | C12'—C11'—H11A | 107.5 |
| C3—C2—C1 | 113.4 (3) | C13'—C11'—H11A | 107.5 |
| C4—C2—H2E | 107.5 | C10'—C11'—H11A | 107.5 |
| C3—C2—H2E | 107.5 | C11'—C12'—H12D | 109.5 |
| C1—C2—H2E | 107.5 | C11'—C12'—H12E | 109.5 |
| C4'—C2'—C3' | 111.1 (3) | H12D—C12'—H12E | 109.5 |
| C4'—C2'—C1' | 109.3 (3) | C11'—C12'—H12F | 109.5 |
| C3'—C2'—C1' | 113.3 (3) | H12D—C12'—H12F | 109.5 |
| C4'—C2'—H2F | 107.7 | H12E—C12'—H12F | 109.5 |
| C3'—C2'—H2F | 107.7 | Na1—O2—H2B | 107 (4) |
| C1'—C2'—H2F | 107.7 | Na1—O2—H2A | 113 (4) |
| C5'—C4'—C9' | 117.3 (4) | H2B—O2—H2A | 116 (7) |
| C5'—C4'—C2' | 121.6 (4) | C11'—C13'—H13D | 109.5 |
| C9'—C4'—C2' | 121.1 (3) | C11'—C13'—H13E | 109.5 |
| C6'—C5'—C4' | 121.5 (4) | H13D—C13'—H13E | 109.5 |
| C6'—C5'—H5A | 119.2 | C11'—C13'—H13F | 109.5 |
| C4'—C5'—H5A | 119.2 | H13D—C13'—H13F | 109.5 |
| C7—C8—C9 | 121.2 (4) | H13E—C13'—H13F | 109.5 |
| C7—C8—H8 | 119.4 | Na1'—O2'—H2D | 110 (5) |
| C9—C8—H8 | 119.4 | Na1'—O2'—H2C | 116 (4) |
| C8'—C9'—C4' | 120.8 (4) | H2D—O2'—H2C | 104 (6) |
| C8'—C9'—H9A | 119.6 | Na1—O1—Na1' | 90.51 (11) |
| C4'—C9'—H9A | 119.6 | Na1—O1—H1A | 137 (4) |
| C4—C9—C8 | 121.0 (4) | Na1'—O1—H1A | 111 (4) |
| C4—C9—H9 | 119.5 | Na1—O1—H1B | 98 (4) |
| C8—C9—H9 | 119.5 | Na1'—O1—H1B | 117 (5) |
| C5—C6—C7 | 121.3 (4) | H1A—O1—H1B | 104 (6) |
| C5—C6—H6 | 119.3 | O4'ii—Na1'—O3' | 101.98 (12) |
| C7—C6—H6 | 119.3 | O4'ii—Na1'—O1 | 164.49 (11) |
| C5'—C6'—C7' | 121.3 (4) | O3'—Na1'—O1 | 86.92 (11) |
| C5'—C6'—H6A | 119.3 | O4'ii—Na1'—O2' | 90.50 (11) |
| C7'—C6'—H6A | 119.3 | O3'—Na1'—O2' | 164.50 (12) |
| C7—C10—C11 | 114.1 (4) | O1—Na1'—O2' | 78.95 (10) |
| C7—C10—H10B | 108.7 | O4'ii—Na1'—O1' | 101.09 (11) |
| C11—C10—H10B | 108.7 | O3'—Na1'—O1' | 101.23 (11) |
| C7—C10—H10A | 108.7 | O1—Na1'—O1' | 89.42 (10) |
| C11—C10—H10A | 108.7 | O2'—Na1'—O1' | 85.11 (10) |
| H10B—C10—H10A | 107.6 | O4'ii—Na1'—Na1 | 145.54 (9) |
| C6—C5—C4 | 120.6 (3) | O3'—Na1'—Na1 | 92.81 (9) |
| C6—C5—H5 | 119.7 | O1—Na1'—Na1 | 44.53 (8) |
| C4—C5—H5 | 119.7 | O2'—Na1'—Na1 | 81.62 (8) |
| C2—C3—H3A | 109.5 | O1'—Na1'—Na1 | 45.04 (7) |
| Symmetry codes: (i) x+1, y, z; (ii) x−1, y, z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H2B···O3′ | 0.85 (2) | 2.15 (2) | 2.990 (4) | 170 (6) |
| O2′—H2D···O3′iii | 0.85 (2) | 1.88 (2) | 2.723 (4) | 173 (7) |
| O1—H1A···O4′iii | 0.85 (2) | 1.92 (2) | 2.766 (4) | 171 (6) |
| O1′—H1C···O4iv | 0.85 (2) | 1.96 (2) | 2.806 (4) | 172 (6) |
| O1′—H1D···O2ii | 0.85 (2) | 1.92 (2) | 2.771 (4) | 177 (6) |
| O2′—H2C···O4 | 0.85 (2) | 2.14 (3) | 2.940 (4) | 158 (6) |
| O2—H2A···O3iv | 0.85 (2) | 1.84 (2) | 2.688 (4) | 176 (6) |
| O1—H1B···O2′i | 0.85 (2) | 1.93 (3) | 2.753 (4) | 162 (6) |
| Symmetry codes: (i) x+1, y, z; (ii) x−1, y, z; (iii) x−1, y+1, z; (iv) x+1, y−1, z. |
Experimental details
| | (I) | (II) |
| Crystal data |
| Chemical formula | Na+·C13H17O2−·2H2O | Na+·C13H17O2−·2H2O |
| Mr | 264.29 | 264.29 |
| Crystal system, space group | Triclinic, P1 | Triclinic, P1 |
| Temperature (K) | 173 | 173 |
| a, b, c (Å) | 5.7396 (4), 6.0284 (4), 23.8301 (17) | 5.7767 (5), 6.0835 (5), 23.880 (2) |
| α, β, γ (°) | 83.457 (1), 89.241 (1), 63.154 (1) | 83.151 (1), 86.482 (1), 61.612 (1) |
| V (Å3) | 730.20 (9) | 733.00 (11) |
| Z | 2 | 2 |
| Radiation type | Mo Kα | Mo Kα |
| µ (mm−1) | 0.11 | 0.11 |
| Crystal size (mm) | 0.40 × 0.30 × 0.10 | 0.30 × 0.30 × 0.10 |
| |
| Data collection |
| Diffractometer | Bruker SMART 1000 CCD area-detector
diffractometer | Bruker SMART 1000 CCD area-detector
diffractometer |
| Absorption correction | Empirical (using intensity measurements)
based on ΔI (SADABS; Bruker, 2000) | Empirical (using intensity measurements)
based on ΔI (SADABS; Bruker, 2000) |
| Tmin, Tmax | 0.791, 1.0 | 0.822, 1.0 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 8727, 3317, 2610 | 7383, 2589, 2350 |
| Rint | 0.037 | 0.033 |
| (sin θ/λ)max (Å−1) | 0.651 | 0.595 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.048, 0.135, 1.05 | 0.040, 0.113, 1.07 |
| No. of reflections | 3317 | 2589 |
| No. of parameters | 185 | 356 |
| No. of restraints | 12 | 37 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.49, −0.43 | 0.57, −0.30 |
Hydrogen-bond geometry (Å, º) for (I) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H2A···O3i | 0.833 (17) | 1.859 (17) | 2.6907 (17) | 176 (3) |
| O1—H1B···O4i | 0.828 (17) | 1.978 (18) | 2.7998 (17) | 172 (3) |
| O1—H1A···O2ii | 0.819 (17) | 1.948 (18) | 2.7507 (17) | 166 (3) |
| O2—H2B···O4iii | 0.831 (17) | 2.042 (18) | 2.8438 (18) | 162 (3) |
| Symmetry codes: (i) x+1, y−1, z; (ii) x+1, y, z; (iii) −x+1, −y, −z. |
Hydrogen-bond geometry (Å, º) for (II) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H2B···O3' | 0.849 (17) | 2.15 (2) | 2.990 (4) | 170 (6) |
| O2'—H2D···O3'i | 0.849 (17) | 1.88 (2) | 2.723 (4) | 173 (7) |
| O1—H1A···O4'i | 0.849 (17) | 1.92 (2) | 2.766 (4) | 171 (6) |
| O1'—H1C···O4ii | 0.849 (17) | 1.96 (2) | 2.806 (4) | 172 (6) |
| O1'—H1D···O2iii | 0.849 (18) | 1.923 (18) | 2.771 (4) | 177 (6) |
| O2'—H2C···O4 | 0.849 (17) | 2.14 (3) | 2.940 (4) | 158 (6) |
| O2—H2A···O3ii | 0.849 (17) | 1.841 (18) | 2.688 (4) | 176 (6) |
| O1—H1B···O2'iv | 0.849 (18) | 1.93 (3) | 2.753 (4) | 162 (6) |
| Symmetry codes: (i) x−1, y+1, z; (ii) x+1, y−1, z; (iii) x−1, y, z; (iv) x+1, y, z. |
&unitsp; top | Racemic | | | | Enantiomeric | | | | |
| Compound name | Space group | Z | Z' | Ref | Space group | Z | Z' | Pseudosymmetry | Ref. |
| 4-Hydroxy-3-(1-phenylpropyl)coumarin | P21/n | 4 | 1 | a | P21 | 4 | 2 | center of symmetry | b |
| 3-[Benzyl(phenyl)phosphinyl]-2-butenoic acid | P21/c | 4 | 1 | c | P21 | 4 | 2 | twofold axis | d |
| Mandelic acid | P21/c | 8 | 2 | e | P21 | 4 | 2 | twofold axis | f |
| {2-[4-(3-Ethoxy-2-hydroxypropoxy)phenylcarbamoyl]ethyl}trimethylammonium p-bromobenzenesulfonate | P1 | 2 | 1 | g | P1 | 2 | 2 | twofold axis | g |
| Ibuprofen sodium dihydrate | P1 | 2 | 1 | h | P1 | 2 | 2 | twofold screw axis | h |
| References: (a) Bravic et al. (1971); (b) Valente et al. (1976); (c) Glówka (1978); (d) Glówka (1981); (e) Fischer & Profir (2003); (f) Patil et al. (1987); (g) Takahashi et al. (2002); (h) this work. |


 journal menu
journal menu metal-organic compounds
metal-organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2005/09/00/sq1215/sq1215fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2005/09/00/sq1215/sq1215fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2005/09/00/sq1215/sq1215fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/c/issues/2005/09/00/sq1215/sq1215fig4thm.gif)




Ibuprofen [α-methyl-4-(isobutyl)phenylacetic acid] is a chiral non-steroidal anti-inflammatory drug (NSAID). The racemic compound, (R/S)-ibuprofen, is usually formulated as tablets or suspensions, which may be purchased without a prescription. The sodium salt of racemic ibuprofen is much more water-soluble than the free acid (Gaikar & Latha, 1997), and crystallizes from water as a dihydrate. Recent publications have described the pharmacokinetics (Soergel et al., 2005), while recent patents have disclosed the formulation of ibuprofen sodium dihydrate (Gruber, 2003; Armitage et al., 1992). This paper compares the crystal structures of racemic [(R/S)-NaIBDH] and enantiomeric [(S)-NaIBDH] ibuprofen sodium dihydrates, (I) and (II), respectively, and explains their similarity.
The ibuprofen sodium dihydrate phase is stable under ambient conditions, while the anhydrous phase is highly hygroscopic. Differential scanning carlorimetry (DSC) and thermogravimetric analysis (TGA) indicate that both structures lose two water molecules in one step. (R/S)-NaIBDH loses 13.5% of weight (theoretical value 13.6%) between 317 and 365 K. The anhydrous (R/S) salt melts at 474 K. (S)-NaIBDH loses 13.1% of weight (theoretical value 13.6%) between 314 and 362 K and the anhydrous (S) salt then melts at 507 K.
The two dihydrate structures are triclinic, with similar lattice parameters and cell volumes. For comparison, the non-reduced cell is shown here for the (S)-NaIBDH structure, while the CIF file with the structure in the reduced cell is provided in the supplementary material [lattice parameters: a = 5.7767 (5), b = 6.0797 (5) and c = 23.880 (2) Å, α = 93.499 (1), β = 93.518 (1) and γ = 118.322 (1)°, cell volume = 733.01 (11) Å3]. The space group of (R/S)-NaIBDH is P1, while that of (S)-NaIBDH is P1. These two structures are very similar in their packing styles, due to the similarity of the coordination interactions. In the (R/S)-NaIBDH structure, two paired R and (S) ibuprofen anions are related by a center of symmetry. The two (S) ibuprofen anions, the two Na+ cations and the four water molecules in the asymmetric unit of (S)-NaIBDH are related by a pseudo twofold screw axis along the a direction. However, the ring formed by the Na+ cations, the water molecules and the carboxylate groups gives the impression of a pseudo center of symmetry. The two structures are shown in Figs. 1 and 2.
The structures clearly show hydrophilic and hydrophobic regions. The hydrophilic regions are formed by the network of water molecules, Na+ cations and carboxylate groups. The carboxylate groups on the ibuprofen molecules are bridged by the Na+ cations to form a one-dimensional infinite chain. In the (R/S)-NaIBDH structure, each of the two chains contains only (R) or (S) molecules, respectively. Two such chains are linked by the water molecules bridging the Na+ cations to form a one-dimensional infinite zipper along the a direction, approximately parallel to the (012) crystallographic plane. Such zippers stack along the b direction and interact by hydrogen bonding to form wafers, the thickness of which is the same as the length of the c dimension, parallel to the ab plane. The two sides of the thick wafers consist of the disordered isobutyl groups and form the hydrophobic regions. The thick wafers stack to form the whole crystal via weak van der Waals interactions.
The water molecule bridging the two Na+ cations donates two lone pairs of electrons, and also donates two H atoms to form hydrogen bonds. The other water molecule donates one lone pair of electrons to an Na+ cation and two H atoms for hydrogen bonding. The interactions surrounding this latter water molecule are strong enough to hinder its loss on a par with the doubly coordinated water molecule, as indicated by the dehydration behavior seen in the TGA. The relatively high stability of the dihydrate phases also explains the hygroscopicity of anhydrous ibuprofen sodium. From the point of view of the hydrogen bonding, two pairs of ibuprofen anions are bridged by two water molecules to form a ring; the rings are bridged by a third water molecule to form a one-dimensional column along the a direction. In (R/S)-NaIBDH, the ring has a centrosymmetric chair conformation, while in (S)-NaIBDH, the ring is not centrosymmetric due to the O2—H2B···O3' hydrogen bonding (Figs. 3 and 4). The hydrogen-bonding geometries of the two structures are provided in Tables 1 and 2.
Each Na+ cation has close contact with three O atoms of the water molecules and two O atoms of two carboxylate groups to form a pyramidal coordination. Two neighboring Na+ cations are bridged by a pair of water O atoms to form a square plane. The Na+ cations are linked by carboxylate groups to form one-dimensional chains. Each carboxylate group coordinates to two Na+ cations and also acts as the proton acceptor for H atoms of three water molecules.
The similarities between the racemic and enantiomeric crystal structures have been observed for other organic molecules, as summarized in Table 3. Comparison shows that one type of structure has two molecules in the asymmetric unit of the enantiomeric structure, which is similar to one pair of asymmetric units in the centrosymmetric structure. In the other type, the unit cell is doubled in the racemic structure and both structures have two molecules in their asymmetric units. Although these structures are similar in packing style, differences arise from the chirality.
This research demonstrates that the dominant interaction for the packing of ibuprofen sodium dihydrate structures is the cation coordination, although the influence of chirality is also seen in the hydrogen-bonding interactions.