Bis(4-picoline-
![[kappa]](/logos/entities/kappa_rmgif.gif) N
N)gold(I) dibromidoaurate(I), [Au(C
6H
7N)
2][AuBr
2], (I), crystallizes in the monoclinic space group
P2
1/
n, with two half cations and one general anion in the asymmetric unit. The cations, located on centres of inversion, assemble to form chains parallel to the
a axis, but there are no significant contacts between the cations. Cohesion is provided by flanking anions, which are connected to the cations by short Au

Au contacts and C-H
Br hydrogen bonds, and to each other by Br
Br contacts. The corresponding chloride derivative, [Au(C
6H
7N)
2][AuCl
2], (II), is isotypic. A previous structure determination of (II), reported in the space group
P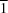
with very similar axis lengths to those of (I) [Lin
et al. (2008).
Inorg. Chem. 47, 2543-2551], might be identical to the structure presented here, except that its
![[gamma]](/logos/entities/gamma_rmgif.gif)
angle of 88.79 (7)° seems to rule out a monoclinic cell. No phase transformation of (II) could be detected on the basis of data sets recorded at 100, 200 and 295 K.
Supporting information
CCDC references: 956978; 956979; 956980; 956981
Compounds (I) (II) were obtained by reaction of 4-picoline with (tht)AuX (tht =
tetrahydrothiophene and X = Br or Cl) [Please give quantities or
mole ratios]. Overlayering the solutions with diethyl ether (X =
Br) or petroleum ether (X = Cl) provided single crystals.
Methyl groups were refined as idealized rigid groups allowed to rotate but not
tip, with C—H = 0.98 Å and H—C—H = 109.5°, and with Uiso(H)
= 1.5Ueq(C). Other H atoms were refined using a riding model starting
from calculated positions, with aromatic C—H = 0.98 Å and Uiso(H)
= 1.2Ueq(C). The methyl H atoms of the room-temperature structure
were indistinct.
For all compounds, data collection: CrysAlis PRO (Agilent, 2013); cell refinement: CrysAlis PRO (Agilent, 2013); data reduction: CrysAlis PRO (Agilent, 2013); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008); molecular graphics: XP (Siemens, 1994); software used to prepare material for publication: SHELXL97 (Sheldrick, 2008).
(I) Bis(4-picoline-
κN)gold(I) dibromidoaurate(I)
top
Crystal data top
| [Au(C6H7N)2][AuBr2] | F(000) = 1312 |
| Mr = 740.01 | Dx = 3.103 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 12461 reflections |
| a = 8.2485 (3) Å | θ = 2.6–30.3° |
| b = 9.7291 (3) Å | µ = 23.53 mm−1 |
| c = 19.8055 (6) Å | T = 100 K |
| β = 94.737 (3)° | Hexagonal tablet, colourless |
| V = 1583.97 (9) Å3 | 0.15 × 0.08 × 0.05 mm |
| Z = 4 | |
Data collection top
Oxford Xcalibur
diffractometer with Eos detector | 4090 independent reflections |
| Radiation source: Enhance (Mo) X-ray Source | 3452 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.072 |
| Detector resolution: 16.1419 pixels mm-1 | θmax = 28.7°, θmin = 2.3° |
| ω scans | h = −11→11 |
Absorption correction: multi-scan
(CrysAlis PRO; Agilent, 2013) | k = −13→13 |
| Tmin = 0.282, Tmax = 1.000 | l = −26→26 |
| 78671 measured reflections | |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.025 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.048 | H-atom parameters constrained |
| S = 1.08 | w = 1/[σ2(Fo2) + (0.0176P)2 + 2.0928P]
where P = (Fo2 + 2Fc2)/3 |
| 4090 reflections | (Δ/σ)max = 0.001 |
| 168 parameters | Δρmax = 1.10 e Å−3 |
| 0 restraints | Δρmin = −1.37 e Å−3 |
Crystal data top
| [Au(C6H7N)2][AuBr2] | V = 1583.97 (9) Å3 |
| Mr = 740.01 | Z = 4 |
| Monoclinic, P21/n | Mo Kα radiation |
| a = 8.2485 (3) Å | µ = 23.53 mm−1 |
| b = 9.7291 (3) Å | T = 100 K |
| c = 19.8055 (6) Å | 0.15 × 0.08 × 0.05 mm |
| β = 94.737 (3)° | |
Data collection top
Oxford Xcalibur
diffractometer with Eos detector | 4090 independent reflections |
Absorption correction: multi-scan
(CrysAlis PRO; Agilent, 2013) | 3452 reflections with I > 2σ(I) |
| Tmin = 0.282, Tmax = 1.000 | Rint = 0.072 |
| 78671 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.025 | 0 restraints |
| wR(F2) = 0.048 | H-atom parameters constrained |
| S = 1.08 | Δρmax = 1.10 e Å−3 |
| 4090 reflections | Δρmin = −1.37 e Å−3 |
| 168 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R-factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Au1 | 0.0000 | 0.5000 | 0.0000 | 0.01804 (6) | |
| Au2 | 0.01325 (2) | 0.730233 (17) | 0.114269 (9) | 0.01520 (5) | |
| Au3 | 0.5000 | 0.5000 | 0.0000 | 0.03044 (8) | |
| Br1 | 0.30111 (6) | 0.73618 (5) | 0.11015 (2) | 0.02044 (10) | |
| Br2 | −0.27195 (6) | 0.73941 (5) | 0.12396 (2) | 0.02214 (11) | |
| N11 | 0.0267 (5) | 0.3507 (4) | 0.07140 (19) | 0.0168 (8) | |
| C12 | 0.1275 (6) | 0.3670 (5) | 0.1275 (2) | 0.0197 (10) | |
| H12 | 0.1904 | 0.4487 | 0.1327 | 0.024* | |
| C13 | 0.1426 (6) | 0.2683 (5) | 0.1780 (2) | 0.0205 (10) | |
| H13 | 0.2133 | 0.2840 | 0.2176 | 0.025* | |
| C14 | 0.0551 (6) | 0.1468 (5) | 0.1711 (2) | 0.0197 (10) | |
| C15 | −0.0461 (6) | 0.1301 (5) | 0.1126 (2) | 0.0224 (11) | |
| H15 | −0.1077 | 0.0480 | 0.1060 | 0.027* | |
| C16 | −0.0588 (6) | 0.2317 (5) | 0.0636 (2) | 0.0211 (10) | |
| H16 | −0.1288 | 0.2179 | 0.0236 | 0.025* | |
| C17 | 0.0678 (7) | 0.0406 (5) | 0.2258 (3) | 0.0325 (13) | |
| H17A | 0.0038 | −0.0403 | 0.2109 | 0.049* | |
| H17B | 0.0260 | 0.0784 | 0.2668 | 0.049* | |
| H17C | 0.1820 | 0.0142 | 0.2356 | 0.049* | |
| N21 | 0.5229 (6) | 0.3462 (4) | 0.0686 (2) | 0.0266 (10) | |
| C22 | 0.4648 (7) | 0.2191 (5) | 0.0534 (3) | 0.0289 (12) | |
| H22 | 0.4156 | 0.2013 | 0.0092 | 0.035* | |
| C23 | 0.4752 (6) | 0.1142 (5) | 0.1008 (2) | 0.0244 (11) | |
| H23 | 0.4331 | 0.0259 | 0.0887 | 0.029* | |
| C24 | 0.5455 (6) | 0.1366 (5) | 0.1650 (2) | 0.0215 (11) | |
| C25 | 0.6075 (7) | 0.2671 (5) | 0.1800 (3) | 0.0256 (11) | |
| H25 | 0.6586 | 0.2859 | 0.2238 | 0.031* | |
| C26 | 0.5950 (7) | 0.3689 (5) | 0.1317 (3) | 0.0278 (12) | |
| H26 | 0.6380 | 0.4574 | 0.1427 | 0.033* | |
| C27 | 0.5548 (7) | 0.0271 (5) | 0.2185 (3) | 0.0288 (12) | |
| H27A | 0.4888 | −0.0519 | 0.2025 | 0.043* | |
| H27B | 0.5135 | 0.0636 | 0.2599 | 0.043* | |
| H27C | 0.6682 | −0.0018 | 0.2281 | 0.043* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Au1 | 0.02986 (15) | 0.01182 (11) | 0.01237 (12) | −0.00069 (10) | 0.00133 (10) | 0.00101 (9) |
| Au2 | 0.01685 (9) | 0.01383 (8) | 0.01463 (8) | −0.00144 (7) | −0.00049 (6) | −0.00024 (7) |
| Au3 | 0.0595 (2) | 0.01621 (13) | 0.01688 (14) | 0.00243 (13) | 0.01097 (13) | 0.00097 (10) |
| Br1 | 0.0165 (2) | 0.0222 (2) | 0.0224 (2) | −0.00326 (18) | 0.00048 (18) | 0.00025 (18) |
| Br2 | 0.0165 (2) | 0.0252 (3) | 0.0244 (3) | −0.00093 (19) | −0.0002 (2) | −0.00162 (19) |
| N11 | 0.026 (2) | 0.0121 (18) | 0.0123 (19) | 0.0002 (16) | 0.0033 (16) | 0.0018 (14) |
| C12 | 0.027 (3) | 0.012 (2) | 0.020 (2) | −0.0009 (19) | 0.001 (2) | −0.0011 (18) |
| C13 | 0.026 (3) | 0.015 (2) | 0.019 (2) | −0.0005 (19) | −0.005 (2) | −0.0025 (18) |
| C14 | 0.028 (3) | 0.018 (2) | 0.013 (2) | 0.004 (2) | 0.002 (2) | −0.0018 (17) |
| C15 | 0.031 (3) | 0.015 (2) | 0.022 (3) | −0.007 (2) | 0.003 (2) | −0.0007 (18) |
| C16 | 0.027 (3) | 0.019 (2) | 0.016 (2) | −0.004 (2) | −0.001 (2) | −0.0025 (18) |
| C17 | 0.054 (4) | 0.019 (3) | 0.024 (3) | −0.007 (2) | −0.006 (3) | 0.008 (2) |
| N21 | 0.049 (3) | 0.015 (2) | 0.018 (2) | 0.0018 (19) | 0.012 (2) | 0.0013 (16) |
| C22 | 0.043 (3) | 0.022 (3) | 0.022 (3) | −0.001 (2) | 0.005 (2) | −0.003 (2) |
| C23 | 0.038 (3) | 0.014 (2) | 0.020 (3) | −0.004 (2) | 0.001 (2) | −0.0052 (18) |
| C24 | 0.032 (3) | 0.014 (2) | 0.020 (2) | 0.004 (2) | 0.007 (2) | 0.0033 (18) |
| C25 | 0.042 (3) | 0.020 (2) | 0.015 (2) | 0.001 (2) | 0.002 (2) | −0.0025 (19) |
| C26 | 0.044 (3) | 0.022 (3) | 0.018 (3) | −0.003 (2) | 0.005 (2) | −0.005 (2) |
| C27 | 0.042 (3) | 0.020 (3) | 0.024 (3) | 0.000 (2) | 0.001 (2) | 0.000 (2) |
Geometric parameters (Å, º) top
| Au1—N11 | 2.027 (4) | C23—C24 | 1.370 (7) |
| Au1—N11i | 2.027 (4) | C24—C25 | 1.391 (7) |
| Au1—Au2i | 3.1796 (2) | C24—C27 | 1.500 (6) |
| Au1—Au2 | 3.1797 (2) | C25—C26 | 1.376 (7) |
| Au2—Br2 | 2.3777 (5) | C12—H12 | 0.9500 |
| Au2—Br1 | 2.3833 (5) | C13—H13 | 0.9500 |
| Au3—N21 | 2.019 (4) | C15—H15 | 0.9500 |
| Au3—N21ii | 2.019 (4) | C16—H16 | 0.9500 |
| N11—C12 | 1.341 (6) | C17—H17A | 0.9800 |
| N11—C16 | 1.357 (6) | C17—H17B | 0.9800 |
| C12—C13 | 1.385 (6) | C17—H17C | 0.9800 |
| C13—C14 | 1.385 (6) | C22—H22 | 0.9500 |
| C14—C15 | 1.381 (7) | C23—H23 | 0.9500 |
| C14—C17 | 1.494 (6) | C25—H25 | 0.9500 |
| C15—C16 | 1.383 (6) | C26—H26 | 0.9500 |
| N21—C22 | 1.351 (6) | C27—H27A | 0.9800 |
| N21—C26 | 1.356 (6) | C27—H27B | 0.9800 |
| C22—C23 | 1.385 (7) | C27—H27C | 0.9800 |
| | | |
| N11—Au1—N11i | 180.0 | N21—C26—C25 | 121.8 (5) |
| N11—Au1—Au2i | 89.25 (10) | N11—C12—H12 | 119.1 |
| N11i—Au1—Au2i | 90.75 (10) | C13—C12—H12 | 119.1 |
| N11—Au1—Au2 | 90.75 (10) | C12—C13—H13 | 119.8 |
| N11i—Au1—Au2 | 89.25 (10) | C14—C13—H13 | 119.8 |
| Au2i—Au1—Au2 | 180.0 | C14—C15—H15 | 119.7 |
| Br2—Au2—Br1 | 175.569 (18) | C16—C15—H15 | 119.7 |
| Br2—Au2—Au1 | 96.191 (13) | N11—C16—H16 | 119.3 |
| Br1—Au2—Au1 | 88.194 (13) | C15—C16—H16 | 119.3 |
| N21—Au3—N21ii | 180.0 | C14—C17—H17A | 109.5 |
| C12—N11—C16 | 118.5 (4) | C14—C17—H17B | 109.5 |
| C12—N11—Au1 | 121.1 (3) | H17A—C17—H17B | 109.5 |
| C16—N11—Au1 | 120.4 (3) | C14—C17—H17C | 109.5 |
| N11—C12—C13 | 121.9 (4) | H17A—C17—H17C | 109.5 |
| C12—C13—C14 | 120.4 (4) | H17B—C17—H17C | 109.5 |
| C15—C14—C13 | 117.2 (4) | N21—C22—H22 | 119.2 |
| C15—C14—C17 | 121.9 (4) | C23—C22—H22 | 119.2 |
| C13—C14—C17 | 120.9 (4) | C24—C23—H23 | 119.7 |
| C14—C15—C16 | 120.7 (4) | C22—C23—H23 | 119.7 |
| N11—C16—C15 | 121.4 (4) | C26—C25—H25 | 119.9 |
| C22—N21—C26 | 118.3 (4) | C24—C25—H25 | 119.9 |
| C22—N21—Au3 | 121.1 (4) | N21—C26—H26 | 119.1 |
| C26—N21—Au3 | 120.6 (3) | C25—C26—H26 | 119.1 |
| N21—C22—C23 | 121.5 (5) | C24—C27—H27A | 109.5 |
| C24—C23—C22 | 120.7 (5) | C24—C27—H27B | 109.5 |
| C23—C24—C25 | 117.6 (4) | H27A—C27—H27B | 109.5 |
| C23—C24—C27 | 122.4 (4) | C24—C27—H27C | 109.5 |
| C25—C24—C27 | 120.0 (5) | H27A—C27—H27C | 109.5 |
| C26—C25—C24 | 120.2 (5) | H27B—C27—H27C | 109.5 |
| | | |
| N11—Au1—Au2—Br2 | −91.17 (11) | C17—C14—C15—C16 | 178.3 (5) |
| N11i—Au1—Au2—Br2 | 88.84 (11) | C12—N11—C16—C15 | 1.4 (7) |
| N11—Au1—Au2—Br1 | 89.47 (11) | Au1—N11—C16—C15 | −178.1 (4) |
| N11i—Au1—Au2—Br1 | −90.53 (11) | C14—C15—C16—N11 | −0.3 (8) |
| Au2i—Au1—N11—C12 | 140.7 (4) | C26—N21—C22—C23 | 1.0 (8) |
| Au2—Au1—N11—C12 | −39.3 (4) | Au3—N21—C22—C23 | −177.8 (4) |
| Au2i—Au1—N11—C16 | −39.8 (3) | N21—C22—C23—C24 | −0.1 (8) |
| Au2—Au1—N11—C16 | 140.2 (3) | C22—C23—C24—C25 | −0.9 (8) |
| C16—N11—C12—C13 | −2.0 (7) | C22—C23—C24—C27 | 178.1 (5) |
| Au1—N11—C12—C13 | 177.5 (4) | C23—C24—C25—C26 | 1.0 (8) |
| N11—C12—C13—C14 | 1.4 (7) | C27—C24—C25—C26 | −178.1 (5) |
| C12—C13—C14—C15 | −0.2 (7) | C22—N21—C26—C25 | −1.0 (8) |
| C12—C13—C14—C17 | −178.8 (5) | Au3—N21—C26—C25 | 177.9 (4) |
| C13—C14—C15—C16 | −0.3 (7) | C24—C25—C26—N21 | 0.0 (8) |
| Symmetry codes: (i) −x, −y+1, −z; (ii) −x+1, −y+1, −z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C12—H12···Br1 | 0.95 | 2.99 | 3.892 (5) | 160 |
| C16—H16···Br1i | 0.95 | 2.94 | 3.847 (5) | 161 |
| C22—H22···Br2i | 0.95 | 2.86 | 3.756 (5) | 158 |
| C26—H26···Br2iii | 0.95 | 2.87 | 3.775 (5) | 159 |
| Symmetry codes: (i) −x, −y+1, −z; (iii) x+1, y, z. |
(II_100K) Bis(4-picoline-
κN)gold(I) dichloridoaurate(I)
top
Crystal data top
| [Au(C6H7N)2][AuCl2] | F(000) = 1168 |
| Mr = 651.09 | Dx = 2.886 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 7483 reflections |
| a = 7.9048 (3) Å | θ = 2.6–30.4° |
| b = 9.5597 (3) Å | µ = 19.90 mm−1 |
| c = 20.1036 (7) Å | T = 100 K |
| β = 99.421 (4)° | Block, colourless |
| V = 1498.68 (9) Å3 | 0.10 × 0.05 × 0.02 mm |
| Z = 4 | |
Data collection top
Oxford Xcalibur
diffractometer with Eos detector | 3726 independent reflections |
| Radiation source: Enhance (Mo) X-ray Source | 3169 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.059 |
| Detector resolution: 16.1419 pixels mm-1 | θmax = 28.3°, θmin = 2.4° |
| w scans | h = −10→10 |
Absorption correction: multi-scan
(CrysAlis PRO; Agilent, 2013) | k = −12→12 |
| Tmin = 0.433, Tmax = 1.000 | l = −26→26 |
| 37223 measured reflections | |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.023 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.040 | H-atom parameters constrained |
| S = 1.04 | w = 1/[σ2(Fo2) + (0.0129P)2 + 0.7856P]
where P = (Fo2 + 2Fc2)/3 |
| 3726 reflections | (Δ/σ)max = 0.004 |
| 168 parameters | Δρmax = 0.86 e Å−3 |
| 0 restraints | Δρmin = −0.92 e Å−3 |
Crystal data top
| [Au(C6H7N)2][AuCl2] | V = 1498.68 (9) Å3 |
| Mr = 651.09 | Z = 4 |
| Monoclinic, P21/n | Mo Kα radiation |
| a = 7.9048 (3) Å | µ = 19.90 mm−1 |
| b = 9.5597 (3) Å | T = 100 K |
| c = 20.1036 (7) Å | 0.10 × 0.05 × 0.02 mm |
| β = 99.421 (4)° | |
Data collection top
Oxford Xcalibur
diffractometer with Eos detector | 3726 independent reflections |
Absorption correction: multi-scan
(CrysAlis PRO; Agilent, 2013) | 3169 reflections with I > 2σ(I) |
| Tmin = 0.433, Tmax = 1.000 | Rint = 0.059 |
| 37223 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.023 | 0 restraints |
| wR(F2) = 0.040 | H-atom parameters constrained |
| S = 1.04 | Δρmax = 0.86 e Å−3 |
| 3726 reflections | Δρmin = −0.92 e Å−3 |
| 168 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R-factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Au1 | 0.0000 | 0.5000 | 0.0000 | 0.01521 (6) | |
| Au2 | 0.01655 (2) | 0.725156 (17) | 0.117825 (9) | 0.01450 (5) | |
| Au3 | 0.5000 | 0.5000 | 0.0000 | 0.02088 (6) | |
| Cl1 | 0.29978 (14) | 0.72685 (12) | 0.11165 (6) | 0.0208 (2) | |
| Cl2 | −0.26357 (14) | 0.73204 (12) | 0.12859 (6) | 0.0214 (2) | |
| N11 | 0.0236 (4) | 0.3462 (4) | 0.06997 (18) | 0.0152 (8) | |
| C12 | 0.1261 (5) | 0.3626 (4) | 0.1300 (2) | 0.0157 (9) | |
| H12 | 0.1957 | 0.4439 | 0.1374 | 0.019* | |
| C13 | 0.1332 (6) | 0.2651 (4) | 0.1809 (2) | 0.0178 (10) | |
| H13 | 0.2053 | 0.2809 | 0.2229 | 0.021* | |
| C14 | 0.0347 (5) | 0.1432 (4) | 0.1710 (2) | 0.0162 (9) | |
| C15 | −0.0651 (5) | 0.1255 (5) | 0.1086 (2) | 0.0180 (10) | |
| H15 | −0.1317 | 0.0429 | 0.0993 | 0.022* | |
| C16 | −0.0690 (5) | 0.2272 (5) | 0.0593 (2) | 0.0172 (9) | |
| H16 | −0.1388 | 0.2128 | 0.0167 | 0.021* | |
| C17 | 0.0321 (6) | 0.0412 (5) | 0.2270 (2) | 0.0251 (11) | |
| H17A | −0.0789 | 0.0462 | 0.2426 | 0.038* | |
| H17B | 0.1239 | 0.0640 | 0.2644 | 0.038* | |
| H17C | 0.0496 | −0.0535 | 0.2108 | 0.038* | |
| N21 | 0.5169 (5) | 0.3414 (4) | 0.06687 (19) | 0.0203 (8) | |
| C22 | 0.4513 (6) | 0.2142 (5) | 0.0486 (2) | 0.0210 (10) | |
| H22 | 0.4015 | 0.1977 | 0.0030 | 0.025* | |
| C23 | 0.4555 (6) | 0.1075 (5) | 0.0952 (2) | 0.0224 (10) | |
| H23 | 0.4071 | 0.0193 | 0.0812 | 0.027* | |
| C24 | 0.5287 (6) | 0.1267 (5) | 0.1621 (2) | 0.0173 (10) | |
| C25 | 0.5994 (6) | 0.2572 (4) | 0.1792 (2) | 0.0178 (10) | |
| H25 | 0.6540 | 0.2747 | 0.2242 | 0.021* | |
| C26 | 0.5910 (6) | 0.3616 (5) | 0.1317 (2) | 0.0206 (10) | |
| H26 | 0.6388 | 0.4506 | 0.1448 | 0.025* | |
| C27 | 0.5290 (6) | 0.0145 (5) | 0.2140 (2) | 0.0228 (11) | |
| H27A | 0.4111 | −0.0040 | 0.2207 | 0.034* | |
| H27B | 0.5974 | 0.0451 | 0.2566 | 0.034* | |
| H27C | 0.5788 | −0.0711 | 0.1985 | 0.034* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Au1 | 0.02321 (13) | 0.01053 (12) | 0.01154 (12) | −0.00073 (9) | 0.00179 (10) | 0.00109 (9) |
| Au2 | 0.01771 (8) | 0.01269 (8) | 0.01244 (8) | −0.00077 (7) | 0.00051 (6) | −0.00074 (7) |
| Au3 | 0.03403 (15) | 0.01536 (13) | 0.01342 (13) | 0.00216 (10) | 0.00437 (11) | 0.00088 (10) |
| Cl1 | 0.0170 (5) | 0.0239 (6) | 0.0207 (6) | −0.0024 (4) | 0.0006 (4) | −0.0004 (5) |
| Cl2 | 0.0183 (5) | 0.0261 (6) | 0.0197 (6) | 0.0003 (4) | 0.0026 (4) | −0.0024 (5) |
| N11 | 0.0199 (19) | 0.0111 (19) | 0.015 (2) | 0.0004 (14) | 0.0035 (16) | −0.0003 (15) |
| C12 | 0.018 (2) | 0.010 (2) | 0.018 (2) | 0.0014 (16) | −0.0005 (19) | −0.0041 (18) |
| C13 | 0.024 (2) | 0.014 (2) | 0.013 (2) | 0.0022 (18) | −0.0013 (19) | −0.0024 (19) |
| C14 | 0.020 (2) | 0.014 (2) | 0.016 (2) | 0.0028 (17) | 0.0065 (19) | 0.0021 (19) |
| C15 | 0.019 (2) | 0.015 (2) | 0.021 (3) | −0.0013 (18) | 0.005 (2) | 0.0010 (19) |
| C16 | 0.022 (2) | 0.015 (2) | 0.014 (2) | −0.0020 (19) | 0.0025 (18) | −0.0022 (19) |
| C17 | 0.034 (3) | 0.018 (2) | 0.022 (3) | −0.003 (2) | 0.001 (2) | 0.004 (2) |
| N21 | 0.029 (2) | 0.015 (2) | 0.017 (2) | 0.0011 (16) | 0.0029 (17) | −0.0006 (16) |
| C22 | 0.030 (3) | 0.017 (2) | 0.014 (2) | 0.001 (2) | −0.001 (2) | −0.005 (2) |
| C23 | 0.030 (3) | 0.016 (2) | 0.021 (3) | −0.003 (2) | 0.000 (2) | −0.001 (2) |
| C24 | 0.022 (2) | 0.016 (2) | 0.015 (2) | 0.0012 (18) | 0.0061 (19) | 0.0000 (19) |
| C25 | 0.022 (2) | 0.018 (3) | 0.013 (2) | 0.0011 (18) | 0.0008 (18) | −0.0024 (18) |
| C26 | 0.028 (3) | 0.017 (2) | 0.018 (3) | −0.0032 (19) | 0.007 (2) | −0.006 (2) |
| C27 | 0.032 (3) | 0.018 (3) | 0.017 (3) | 0.000 (2) | 0.002 (2) | 0.003 (2) |
Geometric parameters (Å, º) top
| Au1—N11i | 2.022 (4) | C23—C24 | 1.385 (6) |
| Au1—N11 | 2.022 (4) | C24—C25 | 1.388 (6) |
| Au1—Au2 | 3.1874 (2) | C24—C27 | 1.496 (6) |
| Au1—Au2i | 3.1875 (2) | C25—C26 | 1.376 (6) |
| Au2—Cl2 | 2.2608 (11) | C12—H12 | 0.9500 |
| Au2—Cl1 | 2.2626 (11) | C13—H13 | 0.9500 |
| Au3—N21 | 2.016 (4) | C15—H15 | 0.9500 |
| Au3—N21ii | 2.016 (4) | C16—H16 | 0.9500 |
| N11—C12 | 1.348 (5) | C17—H17A | 0.9800 |
| N11—C16 | 1.351 (5) | C17—H17B | 0.9800 |
| C12—C13 | 1.379 (6) | C17—H17C | 0.9800 |
| C13—C14 | 1.397 (6) | C22—H22 | 0.9500 |
| C14—C15 | 1.380 (6) | C23—H23 | 0.9500 |
| C14—C17 | 1.492 (6) | C25—H25 | 0.9500 |
| C15—C16 | 1.385 (6) | C26—H26 | 0.9500 |
| N21—C22 | 1.350 (6) | C27—H27A | 0.9800 |
| N21—C26 | 1.352 (6) | C27—H27B | 0.9800 |
| C22—C23 | 1.382 (6) | C27—H27C | 0.9800 |
| | | |
| N11i—Au1—N11 | 180.0 | N21—C26—C25 | 122.0 (4) |
| N11i—Au1—Au2 | 90.79 (10) | N11—C12—H12 | 119.0 |
| N11—Au1—Au2 | 89.21 (10) | C13—C12—H12 | 119.0 |
| N11i—Au1—Au2i | 89.21 (10) | C12—C13—H13 | 119.9 |
| N11—Au1—Au2i | 90.79 (10) | C14—C13—H13 | 119.9 |
| Au2—Au1—Au2i | 180.0 | C14—C15—H15 | 119.8 |
| Cl2—Au2—Cl1 | 176.89 (4) | C16—C15—H15 | 119.8 |
| Cl2—Au2—Au1 | 99.73 (3) | N11—C16—H16 | 119.0 |
| Cl1—Au2—Au1 | 83.38 (3) | C15—C16—H16 | 119.0 |
| N21—Au3—N21ii | 180.0 | C14—C17—H17A | 109.5 |
| C12—N11—C16 | 118.1 (4) | C14—C17—H17B | 109.5 |
| C12—N11—Au1 | 120.7 (3) | H17A—C17—H17B | 109.5 |
| C16—N11—Au1 | 121.1 (3) | C14—C17—H17C | 109.5 |
| N11—C12—C13 | 122.1 (4) | H17A—C17—H17C | 109.5 |
| C12—C13—C14 | 120.3 (4) | H17B—C17—H17C | 109.5 |
| C15—C14—C13 | 117.0 (4) | N21—C22—H22 | 119.5 |
| C15—C14—C17 | 122.0 (4) | C23—C22—H22 | 119.5 |
| C13—C14—C17 | 121.0 (4) | C22—C23—H23 | 119.3 |
| C14—C15—C16 | 120.4 (4) | C24—C23—H23 | 119.3 |
| N11—C16—C15 | 122.0 (4) | C26—C25—H25 | 119.7 |
| C22—N21—C26 | 118.5 (4) | C24—C25—H25 | 119.7 |
| C22—N21—Au3 | 121.0 (3) | N21—C26—H26 | 119.0 |
| C26—N21—Au3 | 120.4 (3) | C25—C26—H26 | 119.0 |
| N21—C22—C23 | 121.0 (4) | C24—C27—H27A | 109.5 |
| C22—C23—C24 | 121.3 (4) | C24—C27—H27B | 109.5 |
| C23—C24—C25 | 116.6 (4) | H27A—C27—H27B | 109.5 |
| C23—C24—C27 | 122.3 (4) | C24—C27—H27C | 109.5 |
| C25—C24—C27 | 121.1 (4) | H27A—C27—H27C | 109.5 |
| C26—C25—C24 | 120.5 (4) | H27B—C27—H27C | 109.5 |
| | | |
| N11i—Au1—Au2—Cl2 | 93.66 (10) | C17—C14—C15—C16 | 175.3 (4) |
| N11—Au1—Au2—Cl2 | −86.34 (10) | C12—N11—C16—C15 | 2.1 (6) |
| N11i—Au1—Au2—Cl1 | −86.39 (10) | Au1—N11—C16—C15 | −174.7 (3) |
| N11—Au1—Au2—Cl1 | 93.61 (10) | C14—C15—C16—N11 | 0.1 (7) |
| Au2—Au1—N11—C12 | −37.4 (3) | C26—N21—C22—C23 | 1.7 (7) |
| Au2i—Au1—N11—C12 | 142.5 (3) | Au3—N21—C22—C23 | −176.8 (3) |
| Au2—Au1—N11—C16 | 139.3 (3) | N21—C22—C23—C24 | −0.8 (7) |
| Au2i—Au1—N11—C16 | −40.7 (3) | C22—C23—C24—C25 | −0.9 (7) |
| C16—N11—C12—C13 | −2.8 (6) | C22—C23—C24—C27 | 177.7 (4) |
| Au1—N11—C12—C13 | 174.1 (3) | C23—C24—C25—C26 | 1.8 (6) |
| N11—C12—C13—C14 | 1.2 (6) | C27—C24—C25—C26 | −176.9 (4) |
| C12—C13—C14—C15 | 1.1 (6) | C22—N21—C26—C25 | −0.8 (7) |
| C12—C13—C14—C17 | −176.0 (4) | Au3—N21—C26—C25 | 177.7 (3) |
| C13—C14—C15—C16 | −1.7 (6) | C24—C25—C26—N21 | −1.0 (7) |
| Symmetry codes: (i) −x, −y+1, −z; (ii) −x+1, −y+1, −z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C12—H12···Cl1 | 0.95 | 2.90 | 3.783 (4) | 155 |
| C16—H16···Cl1i | 0.95 | 2.75 | 3.644 (5) | 158 |
| C17—H17B···Cl1iii | 0.98 | 2.92 | 3.745 (5) | 143 |
| C22—H22···Cl2i | 0.95 | 2.77 | 3.664 (5) | 156 |
| C25—H25···Cl2iii | 0.95 | 2.97 | 3.876 (5) | 159 |
| C26—H26···Cl2iv | 0.95 | 2.83 | 3.727 (5) | 157 |
| C27—H27C···Cl2v | 0.98 | 2.76 | 3.721 (5) | 165 |
| Symmetry codes: (i) −x, −y+1, −z; (iii) −x+1/2, y−1/2, −z+1/2; (iv) x+1, y, z; (v) x+1, y−1, z. |
(II_295K) Bis(4-picolino)gold(I) dichloroaurate(I)
top
Crystal data top
| [Au(C6H7N)2]·[AuCl2] | F(000) = 1168 |
| Mr = 651.09 | Dx = 2.790 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 5451 reflections |
| a = 8.0221 (7) Å | θ = 2.6–21.9° |
| b = 9.7523 (6) Å | µ = 19.23 mm−1 |
| c = 19.9889 (10) Å | T = 295 K |
| β = 97.552 (6)° | Block, colourless |
| V = 1550.26 (18) Å3 | 0.10 × 0.05 × 0.02 mm |
| Z = 4 | |
Data collection top
Oxford Xcalibur
diffractometer with Eos detector | 3283 independent reflections |
| Radiation source: Enhance (Mo) X-ray Source | 2251 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.091 |
| Detector resolution: 16.1419 pixels mm-1 | θmax = 26.7°, θmin = 2.3° |
| ω scans | h = −10→10 |
Absorption correction: multi-scan
(CrysAlis PRO; Agilent, 2013) | k = −12→12 |
| Tmin = 0.392, Tmax = 1.000 | l = −25→25 |
| 48444 measured reflections | |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.035 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.065 | H-atom parameters constrained |
| S = 1.06 | w = 1/[σ2(Fo2) + (0.0217P)2]
where P = (Fo2 + 2Fc2)/3 |
| 3283 reflections | (Δ/σ)max = 0.001 |
| 168 parameters | Δρmax = 0.80 e Å−3 |
| 0 restraints | Δρmin = −0.88 e Å−3 |
Crystal data top
| [Au(C6H7N)2]·[AuCl2] | V = 1550.26 (18) Å3 |
| Mr = 651.09 | Z = 4 |
| Monoclinic, P21/n | Mo Kα radiation |
| a = 8.0221 (7) Å | µ = 19.23 mm−1 |
| b = 9.7523 (6) Å | T = 295 K |
| c = 19.9889 (10) Å | 0.10 × 0.05 × 0.02 mm |
| β = 97.552 (6)° | |
Data collection top
Oxford Xcalibur
diffractometer with Eos detector | 3283 independent reflections |
Absorption correction: multi-scan
(CrysAlis PRO; Agilent, 2013) | 2251 reflections with I > 2σ(I) |
| Tmin = 0.392, Tmax = 1.000 | Rint = 0.091 |
| 48444 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.035 | 0 restraints |
| wR(F2) = 0.065 | H-atom parameters constrained |
| S = 1.06 | Δρmax = 0.80 e Å−3 |
| 3283 reflections | Δρmin = −0.88 e Å−3 |
| 168 parameters | |
Special details top
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are
estimated using the full covariance matrix. The cell esds are taken into
account individually in the estimation of esds in distances, angles and torsion
angles; correlations between esds in cell parameters are only used when they
are defined by crystal symmetry. An approximate (isotropic) treatment of cell
esds is used for estimating esds involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and
goodness of fit S are based on F2, conventional R-factors R are based on F,
with F set to zero for negative F2. The threshold expression of F2 >
2sigma(F2) is used only for calculating R-factors(gt) etc. and is not
relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R-factors based
on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Au1 | 0.0000 | 0.5000 | 0.0000 | 0.05325 (14) | |
| Au2 | 0.00990 (4) | 0.72769 (3) | 0.118601 (15) | 0.05146 (11) | |
| Au3 | 0.5000 | 0.5000 | 0.0000 | 0.07254 (18) | |
| Cl1 | 0.2890 (3) | 0.7347 (2) | 0.11267 (12) | 0.0735 (6) | |
| Cl2 | −0.2669 (3) | 0.7285 (3) | 0.12808 (11) | 0.0759 (7) | |
| N11 | 0.0245 (8) | 0.3499 (6) | 0.0702 (3) | 0.0484 (16) | |
| C12 | 0.1207 (10) | 0.3669 (8) | 0.1300 (4) | 0.053 (2) | |
| H12 | 0.1822 | 0.4475 | 0.1377 | 0.063* | |
| C13 | 0.1315 (10) | 0.2698 (8) | 0.1801 (4) | 0.053 (2) | |
| H13 | 0.1981 | 0.2861 | 0.2210 | 0.064* | |
| C14 | 0.0444 (10) | 0.1483 (8) | 0.1701 (4) | 0.050 (2) | |
| C15 | −0.0490 (10) | 0.1316 (8) | 0.1087 (4) | 0.057 (2) | |
| H15 | −0.1081 | 0.0503 | 0.0995 | 0.068* | |
| C16 | −0.0584 (10) | 0.2315 (8) | 0.0600 (4) | 0.056 (2) | |
| H16 | −0.1242 | 0.2163 | 0.0188 | 0.067* | |
| C17 | 0.0504 (12) | 0.0434 (9) | 0.2262 (4) | 0.078 (3) | |
| H17A | 0.0707 | −0.0458 | 0.2087 | 0.116* | |
| H17B | −0.0549 | 0.0432 | 0.2441 | 0.116* | |
| H17C | 0.1393 | 0.0664 | 0.2614 | 0.116* | |
| N21 | 0.5204 (9) | 0.3433 (7) | 0.0662 (3) | 0.0651 (19) | |
| C22 | 0.4577 (12) | 0.2167 (9) | 0.0492 (4) | 0.072 (3) | |
| H22 | 0.4081 | 0.2011 | 0.0051 | 0.086* | |
| C23 | 0.4653 (11) | 0.1124 (9) | 0.0944 (4) | 0.067 (2) | |
| H23 | 0.4223 | 0.0269 | 0.0807 | 0.081* | |
| C24 | 0.5360 (10) | 0.1321 (9) | 0.1602 (4) | 0.056 (2) | |
| C25 | 0.6011 (11) | 0.2601 (8) | 0.1773 (4) | 0.059 (2) | |
| H25 | 0.6518 | 0.2770 | 0.2211 | 0.071* | |
| C26 | 0.5912 (11) | 0.3625 (9) | 0.1302 (4) | 0.065 (2) | |
| H26 | 0.6350 | 0.4483 | 0.1429 | 0.078* | |
| C27 | 0.5443 (12) | 0.0179 (8) | 0.2125 (4) | 0.080 (3) | |
| H27A | 0.4354 | 0.0056 | 0.2266 | 0.120* | |
| H27B | 0.6240 | 0.0420 | 0.2508 | 0.120* | |
| H27C | 0.5788 | −0.0658 | 0.1931 | 0.120* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Au1 | 0.0820 (4) | 0.0378 (3) | 0.0389 (3) | −0.0009 (2) | 0.0038 (2) | 0.00287 (18) |
| Au2 | 0.0680 (2) | 0.04282 (19) | 0.04147 (18) | −0.00235 (17) | −0.00048 (14) | −0.00302 (14) |
| Au3 | 0.1132 (5) | 0.0569 (3) | 0.0482 (3) | 0.0065 (3) | 0.0130 (3) | 0.0014 (2) |
| Cl1 | 0.0663 (14) | 0.0821 (17) | 0.0696 (14) | −0.0138 (12) | −0.0004 (11) | −0.0026 (12) |
| Cl2 | 0.0674 (15) | 0.0905 (19) | 0.0690 (15) | 0.0030 (12) | 0.0054 (12) | −0.0093 (12) |
| N11 | 0.074 (5) | 0.034 (4) | 0.037 (4) | 0.001 (3) | 0.005 (3) | −0.003 (3) |
| C12 | 0.077 (6) | 0.038 (5) | 0.042 (5) | −0.010 (4) | 0.003 (4) | −0.005 (4) |
| C13 | 0.071 (6) | 0.040 (5) | 0.045 (5) | 0.006 (4) | −0.007 (4) | −0.003 (4) |
| C14 | 0.062 (5) | 0.043 (5) | 0.043 (5) | 0.006 (4) | 0.001 (4) | −0.003 (4) |
| C15 | 0.076 (6) | 0.043 (5) | 0.048 (5) | −0.019 (4) | −0.005 (4) | 0.000 (4) |
| C16 | 0.075 (6) | 0.052 (5) | 0.039 (5) | −0.006 (5) | −0.001 (4) | 0.000 (4) |
| C17 | 0.102 (8) | 0.080 (7) | 0.047 (5) | 0.001 (6) | −0.009 (5) | −0.002 (5) |
| N21 | 0.098 (6) | 0.052 (5) | 0.045 (4) | 0.004 (4) | 0.008 (4) | −0.005 (3) |
| C22 | 0.096 (7) | 0.066 (7) | 0.049 (5) | −0.007 (5) | −0.004 (5) | −0.013 (5) |
| C23 | 0.092 (7) | 0.045 (5) | 0.063 (6) | −0.009 (5) | 0.003 (5) | 0.002 (4) |
| C24 | 0.064 (6) | 0.058 (6) | 0.045 (5) | 0.005 (4) | 0.009 (4) | 0.003 (4) |
| C25 | 0.079 (6) | 0.047 (6) | 0.052 (5) | −0.008 (5) | 0.004 (4) | −0.009 (4) |
| C26 | 0.093 (7) | 0.050 (6) | 0.053 (6) | −0.002 (5) | 0.013 (5) | −0.010 (4) |
| C27 | 0.107 (8) | 0.067 (7) | 0.064 (6) | 0.013 (6) | 0.006 (6) | −0.007 (5) |
Geometric parameters (Å, º) top
| Au1—N11i | 2.019 (6) | C13—C14 | 1.376 (10) |
| Au1—N11 | 2.019 (6) | C14—C15 | 1.362 (9) |
| Au1—Au2i | 3.2415 (3) | C14—C17 | 1.514 (10) |
| Au1—Au2 | 3.2415 (3) | C15—C16 | 1.372 (10) |
| Au2—Cl2 | 2.254 (2) | N21—C26 | 1.342 (9) |
| Au2—Cl1 | 2.259 (2) | N21—C22 | 1.359 (10) |
| Au3—N21 | 2.015 (7) | C22—C23 | 1.356 (11) |
| Au3—N21ii | 2.015 (7) | C23—C24 | 1.376 (10) |
| N11—C16 | 1.335 (9) | C24—C25 | 1.379 (10) |
| N11—C12 | 1.345 (8) | C24—C27 | 1.523 (11) |
| C12—C13 | 1.373 (10) | C25—C26 | 1.368 (10) |
| | | |
| N11i—Au1—N11 | 179.999 (1) | C15—C14—C13 | 116.3 (7) |
| N11i—Au1—Au2i | 89.85 (16) | C15—C14—C17 | 123.2 (7) |
| N11—Au1—Au2i | 90.15 (16) | C13—C14—C17 | 120.5 (7) |
| N11i—Au1—Au2 | 90.15 (16) | C14—C15—C16 | 121.8 (7) |
| N11—Au1—Au2 | 89.85 (16) | N11—C16—C15 | 121.7 (7) |
| Au2i—Au1—Au2 | 180.0 | C26—N21—C22 | 117.5 (7) |
| Cl2—Au2—Cl1 | 177.35 (8) | C26—N21—Au3 | 120.5 (6) |
| Cl2—Au2—Au1 | 97.72 (6) | C22—N21—Au3 | 121.9 (6) |
| Cl1—Au2—Au1 | 84.93 (6) | C23—C22—N21 | 122.1 (8) |
| N21—Au3—N21ii | 179.999 (1) | C22—C23—C24 | 120.6 (8) |
| C16—N11—C12 | 117.3 (6) | C23—C24—C25 | 117.3 (8) |
| C16—N11—Au1 | 121.3 (5) | C23—C24—C27 | 122.0 (8) |
| C12—N11—Au1 | 121.4 (5) | C25—C24—C27 | 120.7 (7) |
| N11—C12—C13 | 122.5 (7) | C26—C25—C24 | 120.2 (8) |
| C12—C13—C14 | 120.4 (7) | N21—C26—C25 | 122.2 (8) |
| | | |
| N11i—Au1—Au2—Cl2 | 92.80 (19) | C17—C14—C15—C16 | 176.7 (8) |
| N11—Au1—Au2—Cl2 | −87.20 (19) | C12—N11—C16—C15 | 1.2 (12) |
| N11i—Au1—Au2—Cl1 | −87.03 (19) | Au1—N11—C16—C15 | −176.7 (6) |
| N11—Au1—Au2—Cl1 | 92.97 (19) | C14—C15—C16—N11 | 0.4 (13) |
| Au2i—Au1—N11—C16 | −38.4 (6) | C26—N21—C22—C23 | 0.0 (13) |
| Au2—Au1—N11—C16 | 141.6 (6) | Au3—N21—C22—C23 | −177.3 (7) |
| Au2i—Au1—N11—C12 | 143.8 (6) | N21—C22—C23—C24 | 0.8 (14) |
| Au2—Au1—N11—C12 | −36.2 (6) | C22—C23—C24—C25 | −1.3 (13) |
| C16—N11—C12—C13 | −1.9 (11) | C22—C23—C24—C27 | 179.1 (8) |
| Au1—N11—C12—C13 | 176.0 (6) | C23—C24—C25—C26 | 1.2 (13) |
| N11—C12—C13—C14 | 1.0 (12) | C27—C24—C25—C26 | −179.2 (8) |
| C12—C13—C14—C15 | 0.6 (12) | C22—N21—C26—C25 | −0.1 (13) |
| C12—C13—C14—C17 | −177.5 (8) | Au3—N21—C26—C25 | 177.2 (7) |
| C13—C14—C15—C16 | −1.3 (12) | C24—C25—C26—N21 | −0.5 (13) |
| Symmetry codes: (i) −x, −y+1, −z; (ii) −x+1, −y+1, −z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C12—H12···Cl1 | 0.93 | 2.99 | 3.864 (8) | 157 |
| C16—H16···Cl1i | 0.93 | 2.82 | 3.710 (8) | 160 |
| C17—H17C···Cl1iii | 0.96 | 3.00 | 3.798 (9) | 142 |
| C22—H22···Cl2i | 0.93 | 2.84 | 3.709 (9) | 157 |
| C26—H26···Cl2iv | 0.93 | 2.87 | 3.748 (9) | 158 |
| C27—H27C···Cl2v | 0.96 | 2.77 | 3.712 (9) | 167 |
| Symmetry codes: (i) −x, −y+1, −z; (iii) −x+1/2, y−1/2, −z+1/2; (iv) x+1, y, z; (v) x+1, y−1, z. |
(II_200K) Bis(4-picolino)gold(I) dichloroaurate(I)
top
Crystal data top
| [Au(C6H7N)2]·[AuCl2] | F(000) = 1168 |
| Mr = 651.09 | Dx = 2.840 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 6856 reflections |
| a = 7.9579 (4) Å | θ = 2.6–26.7° |
| b = 9.6670 (5) Å | µ = 19.58 mm−1 |
| c = 19.9987 (9) Å | T = 200 K |
| β = 98.250 (5)° | Block, colourless |
| V = 1522.55 (13) Å3 | 0.10 × 0.05 × 0.02 mm |
| Z = 4 | |
Data collection top
Oxford Xcalibur
diffractometer with Eos detector | 3929 independent reflections |
| Radiation source: Enhance (Mo) X-ray Source | 2982 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.088 |
| Detector resolution: 16.1419 pixels mm-1 | θmax = 28.7°, θmin = 2.3° |
| ω scans | h = −10→10 |
Absorption correction: multi-scan
(CrysAlis PRO; Agilent, 2013) | k = −13→13 |
| Tmin = 0.412, Tmax = 1.000 | l = −27→27 |
| 57728 measured reflections | |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.031 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.055 | H-atom parameters constrained |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.0184P)2]
where P = (Fo2 + 2Fc2)/3 |
| 3929 reflections | (Δ/σ)max = 0.013 |
| 168 parameters | Δρmax = 0.76 e Å−3 |
| 0 restraints | Δρmin = −0.98 e Å−3 |
Crystal data top
| [Au(C6H7N)2]·[AuCl2] | V = 1522.55 (13) Å3 |
| Mr = 651.09 | Z = 4 |
| Monoclinic, P21/n | Mo Kα radiation |
| a = 7.9579 (4) Å | µ = 19.58 mm−1 |
| b = 9.6670 (5) Å | T = 200 K |
| c = 19.9987 (9) Å | 0.10 × 0.05 × 0.02 mm |
| β = 98.250 (5)° | |
Data collection top
Oxford Xcalibur
diffractometer with Eos detector | 3929 independent reflections |
Absorption correction: multi-scan
(CrysAlis PRO; Agilent, 2013) | 2982 reflections with I > 2σ(I) |
| Tmin = 0.412, Tmax = 1.000 | Rint = 0.088 |
| 57728 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.031 | 0 restraints |
| wR(F2) = 0.055 | H-atom parameters constrained |
| S = 1.05 | Δρmax = 0.76 e Å−3 |
| 3929 reflections | Δρmin = −0.98 e Å−3 |
| 168 parameters | |
Special details top
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are
estimated using the full covariance matrix. The cell esds are taken into
account individually in the estimation of esds in distances, angles and torsion
angles; correlations between esds in cell parameters are only used when they
are defined by crystal symmetry. An approximate (isotropic) treatment of cell
esds is used for estimating esds involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and
goodness of fit S are based on F2, conventional R-factors R are based on F,
with F set to zero for negative F2. The threshold expression of F2 >
2sigma(F2) is used only for calculating R-factors(gt) etc. and is not
relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R-factors based
on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Au1 | 0.0000 | 0.5000 | 0.0000 | 0.03311 (9) | |
| Au2 | 0.01229 (3) | 0.72619 (2) | 0.118343 (11) | 0.03148 (7) | |
| Au3 | 0.5000 | 0.5000 | 0.0000 | 0.04560 (11) | |
| Cl1 | 0.2937 (2) | 0.73117 (17) | 0.11232 (8) | 0.0444 (4) | |
| Cl2 | −0.26575 (19) | 0.72982 (18) | 0.12844 (8) | 0.0458 (4) | |
| N11 | 0.0240 (6) | 0.3469 (5) | 0.0704 (2) | 0.0310 (11) | |
| C12 | 0.1225 (7) | 0.3640 (6) | 0.1300 (3) | 0.0343 (14) | |
| H12 | 0.1880 | 0.4462 | 0.1375 | 0.041* | |
| C13 | 0.1324 (7) | 0.2674 (6) | 0.1806 (3) | 0.0335 (13) | |
| H13 | 0.2028 | 0.2838 | 0.2224 | 0.040* | |
| C14 | 0.0403 (7) | 0.1461 (6) | 0.1710 (3) | 0.0310 (13) | |
| C15 | −0.0559 (8) | 0.1276 (6) | 0.1087 (3) | 0.0380 (15) | |
| H15 | −0.1181 | 0.0442 | 0.0993 | 0.046* | |
| C16 | −0.0628 (7) | 0.2291 (6) | 0.0595 (3) | 0.0327 (13) | |
| H16 | −0.1309 | 0.2145 | 0.0171 | 0.039* | |
| C17 | 0.0427 (9) | 0.0414 (7) | 0.2266 (3) | 0.0489 (17) | |
| H17A | −0.0574 | 0.0542 | 0.2493 | 0.073* | |
| H17B | 0.1457 | 0.0536 | 0.2593 | 0.073* | |
| H17C | 0.0413 | −0.0519 | 0.2074 | 0.073* | |
| N21 | 0.5190 (6) | 0.3429 (5) | 0.0667 (2) | 0.0403 (12) | |
| C22 | 0.4541 (8) | 0.2165 (7) | 0.0488 (3) | 0.0453 (16) | |
| H22 | 0.4034 | 0.2008 | 0.0035 | 0.054* | |
| C23 | 0.4601 (8) | 0.1101 (6) | 0.0950 (3) | 0.0409 (15) | |
| H23 | 0.4134 | 0.0225 | 0.0811 | 0.049* | |
| C24 | 0.5332 (7) | 0.1295 (6) | 0.1612 (3) | 0.0350 (14) | |
| C25 | 0.6015 (8) | 0.2587 (6) | 0.1781 (3) | 0.0386 (15) | |
| H25 | 0.6552 | 0.2758 | 0.2229 | 0.046* | |
| C26 | 0.5923 (8) | 0.3614 (7) | 0.1312 (3) | 0.0421 (16) | |
| H26 | 0.6393 | 0.4493 | 0.1444 | 0.051* | |
| C27 | 0.5368 (9) | 0.0156 (6) | 0.2137 (3) | 0.0467 (17) | |
| H27A | 0.4206 | −0.0154 | 0.2163 | 0.070* | |
| H27B | 0.5880 | 0.0511 | 0.2579 | 0.070* | |
| H27C | 0.6040 | −0.0624 | 0.2009 | 0.070* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Au1 | 0.0510 (2) | 0.02356 (17) | 0.02408 (17) | −0.00100 (14) | 0.00290 (14) | 0.00204 (13) |
| Au2 | 0.03985 (13) | 0.02709 (12) | 0.02623 (11) | −0.00144 (11) | 0.00044 (9) | −0.00162 (10) |
| Au3 | 0.0726 (3) | 0.0346 (2) | 0.03005 (19) | 0.00465 (18) | 0.00897 (18) | 0.00138 (15) |
| Cl1 | 0.0388 (8) | 0.0506 (10) | 0.0422 (9) | −0.0081 (7) | 0.0002 (7) | −0.0011 (7) |
| Cl2 | 0.0401 (8) | 0.0557 (11) | 0.0409 (9) | 0.0009 (7) | 0.0033 (7) | −0.0057 (8) |
| N11 | 0.043 (3) | 0.026 (3) | 0.023 (2) | 0.002 (2) | 0.004 (2) | 0.001 (2) |
| C12 | 0.047 (4) | 0.022 (3) | 0.034 (3) | 0.000 (3) | 0.006 (3) | −0.005 (3) |
| C13 | 0.042 (3) | 0.028 (3) | 0.028 (3) | 0.005 (3) | −0.004 (3) | 0.000 (2) |
| C14 | 0.043 (3) | 0.022 (3) | 0.028 (3) | 0.000 (3) | 0.007 (3) | −0.003 (2) |
| C15 | 0.050 (4) | 0.030 (4) | 0.032 (3) | −0.011 (3) | 0.001 (3) | −0.001 (3) |
| C16 | 0.044 (3) | 0.030 (3) | 0.023 (3) | −0.006 (3) | 0.000 (3) | 0.002 (2) |
| C17 | 0.062 (4) | 0.046 (4) | 0.035 (4) | 0.001 (3) | −0.003 (3) | 0.007 (3) |
| N21 | 0.060 (3) | 0.029 (3) | 0.032 (3) | 0.004 (3) | 0.009 (3) | −0.003 (2) |
| C22 | 0.062 (4) | 0.041 (4) | 0.032 (3) | −0.006 (3) | 0.005 (3) | −0.007 (3) |
| C23 | 0.057 (4) | 0.031 (4) | 0.034 (3) | −0.005 (3) | 0.003 (3) | −0.007 (3) |
| C24 | 0.039 (3) | 0.036 (4) | 0.031 (3) | 0.002 (3) | 0.009 (3) | −0.004 (3) |
| C25 | 0.052 (4) | 0.034 (4) | 0.029 (3) | 0.004 (3) | 0.005 (3) | −0.005 (3) |
| C26 | 0.057 (4) | 0.031 (4) | 0.038 (4) | −0.005 (3) | 0.009 (3) | −0.010 (3) |
| C27 | 0.065 (5) | 0.029 (4) | 0.043 (4) | −0.002 (3) | −0.001 (3) | −0.004 (3) |
Geometric parameters (Å, º) top
| Au1—N11 | 2.032 (4) | C13—C14 | 1.381 (7) |
| Au1—N11i | 2.033 (4) | C14—C15 | 1.378 (7) |
| Au1—Au2i | 3.2133 (2) | C14—C17 | 1.501 (8) |
| Au1—Au2 | 3.2134 (2) | C15—C16 | 1.385 (7) |
| Au2—Cl2 | 2.2507 (15) | N21—C26 | 1.350 (7) |
| Au2—Cl1 | 2.2602 (16) | N21—C22 | 1.355 (7) |
| Au3—N21 | 2.012 (5) | C22—C23 | 1.378 (8) |
| Au3—N21ii | 2.012 (5) | C23—C24 | 1.380 (8) |
| N11—C16 | 1.334 (7) | C24—C25 | 1.385 (8) |
| N11—C12 | 1.340 (7) | C24—C27 | 1.520 (8) |
| C12—C13 | 1.371 (8) | C25—C26 | 1.360 (8) |
| | | |
| N11—Au1—N11i | 180.0 (3) | C15—C14—C13 | 116.9 (5) |
| N11—Au1—Au2i | 90.27 (12) | C15—C14—C17 | 121.9 (5) |
| N11i—Au1—Au2i | 89.73 (12) | C13—C14—C17 | 121.2 (5) |
| N11—Au1—Au2 | 89.73 (12) | C14—C15—C16 | 120.7 (5) |
| N11i—Au1—Au2 | 90.27 (12) | N11—C16—C15 | 121.5 (5) |
| Au2i—Au1—Au2 | 180.0 | C26—N21—C22 | 117.9 (5) |
| Cl2—Au2—Cl1 | 177.04 (6) | C26—N21—Au3 | 121.0 (4) |
| Cl2—Au2—Au1 | 98.64 (4) | C22—N21—Au3 | 121.1 (4) |
| Cl1—Au2—Au1 | 84.31 (4) | N21—C22—C23 | 121.5 (6) |
| N21—Au3—N21ii | 179.998 (1) | C22—C23—C24 | 120.6 (6) |
| C16—N11—C12 | 118.3 (5) | C23—C24—C25 | 117.0 (6) |
| C16—N11—Au1 | 120.9 (4) | C23—C24—C27 | 121.9 (6) |
| C12—N11—Au1 | 120.8 (4) | C25—C24—C27 | 121.1 (5) |
| N11—C12—C13 | 122.5 (5) | C26—C25—C24 | 120.6 (6) |
| C12—C13—C14 | 120.1 (5) | N21—C26—C25 | 122.4 (6) |
| | | |
| N11—Au1—Au2—Cl2 | −86.86 (13) | C17—C14—C15—C16 | 176.6 (6) |
| N11i—Au1—Au2—Cl2 | 93.14 (13) | C12—N11—C16—C15 | 1.6 (8) |
| N11—Au1—Au2—Cl1 | 93.26 (13) | Au1—N11—C16—C15 | −175.9 (4) |
| N11i—Au1—Au2—Cl1 | −86.74 (13) | C14—C15—C16—N11 | 0.7 (9) |
| Au2i—Au1—N11—C16 | −39.3 (4) | C26—N21—C22—C23 | 0.9 (9) |
| Au2—Au1—N11—C16 | 140.7 (4) | Au3—N21—C22—C23 | −177.2 (5) |
| Au2i—Au1—N11—C12 | 143.3 (4) | N21—C22—C23—C24 | −0.1 (10) |
| Au2—Au1—N11—C12 | −36.7 (4) | C22—C23—C24—C25 | −1.0 (9) |
| C16—N11—C12—C13 | −2.3 (8) | C22—C23—C24—C27 | 178.0 (6) |
| Au1—N11—C12—C13 | 175.2 (4) | C23—C24—C25—C26 | 1.4 (9) |
| N11—C12—C13—C14 | 0.7 (9) | C27—C24—C25—C26 | −177.7 (6) |
| C12—C13—C14—C15 | 1.5 (9) | C22—N21—C26—C25 | −0.6 (9) |
| C12—C13—C14—C17 | −177.3 (6) | Au3—N21—C26—C25 | 177.5 (5) |
| C13—C14—C15—C16 | −2.2 (9) | C24—C25—C26—N21 | −0.6 (10) |
| Symmetry codes: (i) −x, −y+1, −z; (ii) −x+1, −y+1, −z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C12—H12···Cl1 | 0.95 | 2.94 | 3.836 (6) | 157 |
| C16—H16···Cl1i | 0.95 | 2.77 | 3.677 (6) | 159 |
| C22—H22···Cl2i | 0.95 | 2.79 | 3.682 (6) | 157 |
| C25—H25···Cl2iii | 0.95 | 3.01 | 3.912 (6) | 159 |
| C26—H26···Cl2iv | 0.95 | 2.85 | 3.739 (7) | 157 |
| C27—H27C···Cl2v | 0.98 | 2.76 | 3.712 (7) | 163 |
| Symmetry codes: (i) −x, −y+1, −z; (iii) −x+1/2, y−1/2, −z+1/2; (iv) x+1, y, z; (v) x+1, y−1, z. |
Experimental details
| | (I) | (II_100K) | (II_295K) | (II_200K) |
| Crystal data |
| Chemical formula | [Au(C6H7N)2][AuBr2] | [Au(C6H7N)2][AuCl2] | [Au(C6H7N)2]·[AuCl2] | [Au(C6H7N)2]·[AuCl2] |
| Mr | 740.01 | 651.09 | 651.09 | 651.09 |
| Crystal system, space group | Monoclinic, P21/n | Monoclinic, P21/n | Monoclinic, P21/n | Monoclinic, P21/n |
| Temperature (K) | 100 | 100 | 295 | 200 |
| a, b, c (Å) | 8.2485 (3), 9.7291 (3), 19.8055 (6) | 7.9048 (3), 9.5597 (3), 20.1036 (7) | 8.0221 (7), 9.7523 (6), 19.9889 (10) | 7.9579 (4), 9.6670 (5), 19.9987 (9) |
| β (°) | 94.737 (3) | 99.421 (4) | 97.552 (6) | 98.250 (5) |
| V (Å3) | 1583.97 (9) | 1498.68 (9) | 1550.26 (18) | 1522.55 (13) |
| Z | 4 | 4 | 4 | 4 |
| Radiation type | Mo Kα | Mo Kα | Mo Kα | Mo Kα |
| µ (mm−1) | 23.53 | 19.90 | 19.23 | 19.58 |
| Crystal size (mm) | 0.15 × 0.08 × 0.05 | 0.10 × 0.05 × 0.02 | 0.10 × 0.05 × 0.02 | 0.10 × 0.05 × 0.02 |
| |
| Data collection |
| Diffractometer | Oxford Xcalibur
diffractometer with Eos detector | Oxford Xcalibur
diffractometer with Eos detector | Oxford Xcalibur
diffractometer with Eos detector | Oxford Xcalibur
diffractometer with Eos detector |
| Absorption correction | Multi-scan
(CrysAlis PRO; Agilent, 2013) | Multi-scan
(CrysAlis PRO; Agilent, 2013) | Multi-scan
(CrysAlis PRO; Agilent, 2013) | Multi-scan
(CrysAlis PRO; Agilent, 2013) |
| Tmin, Tmax | 0.282, 1.000 | 0.433, 1.000 | 0.392, 1.000 | 0.412, 1.000 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 78671, 4090, 3452 | 37223, 3726, 3169 | 48444, 3283, 2251 | 57728, 3929, 2982 |
| Rint | 0.072 | 0.059 | 0.091 | 0.088 |
| (sin θ/λ)max (Å−1) | 0.676 | 0.667 | 0.633 | 0.676 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.025, 0.048, 1.08 | 0.023, 0.040, 1.04 | 0.035, 0.065, 1.06 | 0.031, 0.055, 1.05 |
| No. of reflections | 4090 | 3726 | 3283 | 3929 |
| No. of parameters | 168 | 168 | 168 | 168 |
| H-atom treatment | H-atom parameters constrained | H-atom parameters constrained | H-atom parameters constrained | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 1.10, −1.37 | 0.86, −0.92 | 0.80, −0.88 | 0.76, −0.98 |
Hydrogen-bond geometry (Å, º) for (I) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C12—H12···Br1 | 0.95 | 2.99 | 3.892 (5) | 160 |
| C16—H16···Br1i | 0.95 | 2.94 | 3.847 (5) | 161 |
| C22—H22···Br2i | 0.95 | 2.86 | 3.756 (5) | 158 |
| C26—H26···Br2ii | 0.95 | 2.87 | 3.775 (5) | 159 |
| Symmetry codes: (i) −x, −y+1, −z; (ii) x+1, y, z. |
Hydrogen-bond geometry (Å, º) for (II_100K) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C12—H12···Cl1 | 0.95 | 2.90 | 3.783 (4) | 155 |
| C16—H16···Cl1i | 0.95 | 2.75 | 3.644 (5) | 158 |
| C22—H22···Cl2i | 0.95 | 2.77 | 3.664 (5) | 156 |
| C26—H26···Cl2ii | 0.95 | 2.83 | 3.727 (5) | 157 |
| Symmetry codes: (i) −x, −y+1, −z; (ii) x+1, y, z. |
Hydrogen-bond geometry (Å, º) for (II_295K) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C12—H12···Cl1 | 0.93 | 2.99 | 3.864 (8) | 157 |
| C16—H16···Cl1i | 0.93 | 2.82 | 3.710 (8) | 160 |
| C17—H17C···Cl1ii | 0.96 | 3.00 | 3.798 (9) | 142 |
| C22—H22···Cl2i | 0.93 | 2.84 | 3.709 (9) | 157 |
| C26—H26···Cl2iii | 0.93 | 2.87 | 3.748 (9) | 158 |
| C27—H27C···Cl2iv | 0.96 | 2.77 | 3.712 (9) | 167 |
| Symmetry codes: (i) −x, −y+1, −z; (ii) −x+1/2, y−1/2, −z+1/2; (iii) x+1, y, z; (iv) x+1, y−1, z. |
Hydrogen-bond geometry (Å, º) for (II_200K) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C12—H12···Cl1 | 0.95 | 2.94 | 3.836 (6) | 157 |
| C16—H16···Cl1i | 0.95 | 2.77 | 3.677 (6) | 159 |
| C22—H22···Cl2i | 0.95 | 2.79 | 3.682 (6) | 157 |
| C25—H25···Cl2ii | 0.95 | 3.01 | 3.912 (6) | 159 |
| C26—H26···Cl2iii | 0.95 | 2.85 | 3.739 (7) | 157 |
| C27—H27C···Cl2iv | 0.98 | 2.76 | 3.712 (7) | 163 |
| Symmetry codes: (i) −x, −y+1, −z; (ii) −x+1/2, y−1/2, −z+1/2; (iii) x+1, y, z; (iv) x+1, y−1, z. |
Selected geometric parameters (Å, °) for (I) and (II) top| Bond/angle | (I) (X = Br) | (II) (X = Cl) |
| Au1—N11 | 2.027 (4) | 2.022 (4) |
| Au2—Br1 | 2.3833 (5) | 2.2626 (11) |
| Au2—Br2 | 2.3777 (5) | 2.2608 (11) |
| Au3—N21 | 2.019 (4) | 2.016 (4) |
| | |
| Br1—Au2—Br2 | 175.569 (18) | 176.89 (4) |


 journal menu
journal menu metal-organic compounds
metal-organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2013/07/00/sk3492/sk3492fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2013/07/00/sk3492/sk3492fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2013/07/00/sk3492/sk3492fig3thm.gif)




Complexes of gold(I) halides with pyridines, picolines and lutidines L are generally of the form (L2Au)+.(AuX2)-, whereby the ions are linked by Au···Au contacts to form finite or infinite chains. The pioneering work on these complexes was performed by Strähle and co-workers (see e.g. Adams et al., 1982; Conzelmann et al., 1984). However, the complex (2-picoline)AuCl is molecular (Jones & Ahrens, 1998).
We are interested in amine complexes of gold (Döring & Jones, 2013a, and references therein) and have recently begun to study amine complexes of gold(I) bromide (Döring & Jones, 2013b). We began by determining the structure of the 4-picoline complex of AuBr, (I), which adopts the usual ionic form. A comparison with the structure of the analogous chloro derivative, (II) (Lin et al., 2008), revealed some apparent inconsistencies, which led us to redetermine the latter structure.
Compound (I) crystallizes in space group P21/n with Z = 4. The asymmetric unit contains two half cations, in which the Au atoms occupy inversion centres, and a complete anion (Fig. 1). Fig. 2 shows the packing of the various ions. The cations assemble to form chains parallel to the a axis; the cations based on Au1 and Au3 occupy alternate positions, and the N—Au—N vectors are perpendicular to the chain direction. The Au1···Au3 distances within the chain are, at 4.1243 (2) Å, too long to represent significant aurophilic interactions.
The cations centred on Au1 are flanked by the anions via a short contact Au1···Au2 = 3.1796 (2) Å. Further support for the chain is provided by four `weak' hydrogen bonds of C—H···Br type (Table 2) involving the ortho H atoms of the picoline ligands. We have previously drawn attention to the importance of such interactions in the packing of bis(3-bromopyridine)gold(I) dichloroaurate(I) (Freytag & Jones, 2000), and thus in general for other gold complexes with pyridine-type ligands. Fig. 2 also shows the short Br···Br contacts of 3.5098 (7) Å between neighbouring anions. These are the shortest known between AuBr2- anions, the previous shortest according to a search of the Cambridge Structural Database (CSD, Version?; Allen, 2002) being 3.677 Å in a tetrathiafulvalene charge-transfer complex (Beno et al., 1990), but they are probably too linear [Au—Br···Br = 173.53 (2) and 170.55 (2)°] to be regarded as significant stabilizing interactions (Pedireddi et al., 1994). Similarly, Au3···Br contacts of 3.6–3.7 Å are also probably too long to be significant, and are not shown in Fig. 2.
The structure of (II), as published by Lin et al. (2008), was reported in space group P1 with cell constants (at room temperature) a = 8.121 (8), b = 9.827 (7) and c = 19.982 (7) Å, α = 89.72 (4), β = 82.21 (4) and γ = 88.79 (7), and Z = 4 (four independent cations with inversion symmetry and two independent anions on general positions). General features of the packing were closely analogous to those of (I). In general, analogous chloro and bromo derivatives may well be isotypic. The general similarity of the cell of (II) to that of (I) is clear, if the complementary angle for β is used, but the value of γ seems to rule out a monoclinic setting for (II). We therefore decided to redetermine the structure of (II) at low temperature.
The structure of (II) at 100 K is shown in Fig. 3. Compounds (I) and (II) are indeed isotypic at 100 K. There are significant shifts in the cell constants, especially β, but the coordinates of (I) can be used as a starting point for the refinement of (II) and vice versa. Details of C—H···Cl hydrogen bonds within the chains are given in Table 3. Further borderline contacts between chains, not observed for the bromo derivative, are given in the Supplementary Materials. Contact distances: Au1···Au2 = 3.1874 (2), Au1···Au3 = 3.9524 (2) and Cl1···Cl2 between anions = 3.4128 (14) Å. A database search revealed no other structures with short Cl···Cl contacts between AuCl2- anions. No separate packing diagram is presented because of the close similarity to the packing of (I).
If the structure of (II) is indeed monoclinic at 100 K but triclinic at room temperature, presumably a phase transition must take place somewhere between these two temperatures. We therefore remeasured the structure at 200 and 295 K. However, the monoclinic cell remained essentially unchanged, apart from slight changes in cell constants, especially the β angle [at 200 K, a = 7.9579 (4), b = 9.6670 (5) and c = 19.9987 (9) Å, β = 98.250 (5)° and V = 1522.55 (13) Å3; at 295 K, a = 8.0221 (7), b = 9.7523 (6) and c = 19.9889 (19) Å, β = 97.552 (6)° and V = 1550.26 (18) Å3], corresponding to the expected increase in cell volume with temperature. In particular, there was no evidence for a significant deviation of α or γ from 90°. Details of these structures are contained in the deposited CIF files.
Finally, we used the deposited structure of (II) (Lin et al., 2008) to calculate structure factors (using the program XPREP, which forms part of the SHELX suite; Sheldrick, 2008). The same program was then used to check for higher symmetry, increasing the angle tolerance to 2°. The program chose a monoclinic cell [space group P21/n, same axes, α = 89.72, β = 97.79 and γ = 91.21°, matrix 1 0 0 / 0 1 0 / 0 0 1] with an R(int) value of 0.02, and the intensity data thus transformed could be used to refine our structure of (II) with a simulated R value of essentially zero. It therefore seems possible that the two structures are identical, and that errors in the apparent values for the α and γ angles may have led to the false assumption of triclinic symmetry for (II). Clearly, the crystal quality was only moderate, in view of the reported wR2 value of 0.218. We too were once misled in one such case when the preliminary cell from a poor-quality crystal had an angle just greater than 91°; the program decided on a triclinic cell and, in the pre-CheckCIF era, we carelessly failed to check this assumption, leading to the publication of a structure in an incorrect space group (Ahrens et al., 2000; CSD refcode DUHQUS, later corrected as DUHQUS01, space group C2/c). However, we cannot with total certainty rule out the coexistence of two closely similar structures, one triclinic and one monoclinic. The powder patterns would be almost identical, and the compound is not especially stable, so extensive attempts to interconvert putative polymorphs would probably be unsuccessful.
We contacted the starred author of the previous report but he was unable to comment for health reasons.