In the polymeric title compound, [CuCl
2(C
6H
6N
4)]
n, each Cu
II ion is five-coordinated by four basal atoms (two N atoms from a 2,2'-biimidazole molecule and two Cl
- anions) and one axial Cl
- anion, in a distorted square-pyramidal coordination geometry. Cl
- anions bridge the {Cu(C
6H
6N
4)Cl} units into one-dimensional linear chains, which are reinforced by
![[pi]](/logos/entities/pi_rmgif.gif)
-
interactions. Adjacent linear chains are linked by N-H

Cl hydrogen bonds, resulting in a grid layer. The hydrogen-bonding pattern can be described in graph-set notation as
C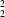
(9)
R(9)
R(14). This study extends our knowledge of the multifunctional properties of the 2,2'-biimidazole ligand and of the coordination stereochemistry of copper(II).
Supporting information
CCDC reference: 285638
CuCl2·2H2O (1 mmol, 0.17 g) and 2,2'-biimidazole (1 mmol, 0.14 g) were suspended in water (30 ml). To the resulting mixture, concentrated aqueous HCl was added until the suspension became clear. The resulting solution was filtered and allowed to evaporate slowly at room temperature. After three weeks, blue crystals of (I) appeared.
All H atoms were positioned geometrically and allowed to ride on their parent atoms at distances of C—H = 0.93 Å or N—H = 0.86 Å, and with Uiso(H) = 1.2Ueq(parent atom).
Data collection: SMART (Bruker, 2002); cell refinement: SAINT (Bruker, 2002); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: SHELXTL (Bruker, 2002); software used to prepare material for publication: SHELXL97.
catena-Poly[[(2,2'-biimidazole-
κ2N,
N')chlorocopper(II)]-µ-chloro]
top
Crystal data top
| [CuCl2(C6H6N4)] | F(000) = 266 |
| Mr = 268.59 | Dx = 2.032 Mg m−3 |
| Monoclinic, P21 | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: P 2yb | Cell parameters from 1408 reflections |
| a = 3.8671 (4) Å | θ = 2.7–24.2° |
| b = 14.8937 (17) Å | µ = 3.05 mm−1 |
| c = 7.6363 (9) Å | T = 298 K |
| β = 93.742 (2)° | Needle, blue |
| V = 438.88 (9) Å3 | 0.34 × 0.06 × 0.05 mm |
| Z = 2 | |
Data collection top
Bruker APEX area-detector
diffractometer | 1590 independent reflections |
| Radiation source: fine-focus sealed tube | 1522 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.038 |
| ϕ and ω scans | θmax = 25.2°, θmin = 2.7° |
Absorption correction: multi-scan
(SADABS; Bruker, 2002) | h = −4→4 |
| Tmin = 0.424, Tmax = 0.863 | k = −17→17 |
| 4178 measured reflections | l = −9→9 |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.053 | H-atom parameters constrained |
| wR(F2) = 0.103 | w = 1/[σ2(Fo2) + (0.0324P)2 + 0.9884P]
where P = (Fo2 + 2Fc2)/3 |
| S = 1.19 | (Δ/σ)max < 0.001 |
| 1590 reflections | Δρmax = 0.70 e Å−3 |
| 118 parameters | Δρmin = −0.76 e Å−3 |
| 1 restraint | Absolute structure: Flack (1983), with 761 Friedel pairs |
| Primary atom site location: structure-invariant direct methods | Absolute structure parameter: 0.07 (3) |
Crystal data top
| [CuCl2(C6H6N4)] | V = 438.88 (9) Å3 |
| Mr = 268.59 | Z = 2 |
| Monoclinic, P21 | Mo Kα radiation |
| a = 3.8671 (4) Å | µ = 3.05 mm−1 |
| b = 14.8937 (17) Å | T = 298 K |
| c = 7.6363 (9) Å | 0.34 × 0.06 × 0.05 mm |
| β = 93.742 (2)° | |
Data collection top
Bruker APEX area-detector
diffractometer | 1590 independent reflections |
Absorption correction: multi-scan
(SADABS; Bruker, 2002) | 1522 reflections with I > 2σ(I) |
| Tmin = 0.424, Tmax = 0.863 | Rint = 0.038 |
| 4178 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.053 | H-atom parameters constrained |
| wR(F2) = 0.103 | Δρmax = 0.70 e Å−3 |
| S = 1.19 | Δρmin = −0.76 e Å−3 |
| 1590 reflections | Absolute structure: Flack (1983), with 761 Friedel pairs |
| 118 parameters | Absolute structure parameter: 0.07 (3) |
| 1 restraint | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Cu1 | 0.3637 (2) | 0.86139 (5) | 0.63901 (10) | 0.0233 (2) | |
| Cl1 | 0.3741 (5) | 0.75984 (13) | 0.8595 (2) | 0.0297 (5) | |
| Cl2 | 0.7743 (5) | 0.95448 (14) | 0.7671 (2) | 0.0300 (5) | |
| N1 | 0.0701 (15) | 0.7804 (4) | 0.4796 (8) | 0.0204 (13) | |
| N2 | −0.1181 (16) | 0.7476 (4) | 0.2112 (9) | 0.0285 (15) | |
| H2A | −0.1604 | 0.7514 | 0.0995 | 0.034* | |
| N3 | 0.2332 (18) | 0.9406 (4) | 0.1335 (8) | 0.0317 (16) | |
| H3A | 0.1502 | 0.9267 | 0.0299 | 0.038* | |
| N4 | 0.3698 (16) | 0.9325 (4) | 0.4157 (8) | 0.0246 (14) | |
| C1 | −0.0925 (18) | 0.6984 (5) | 0.4822 (10) | 0.0256 (18) | |
| H1 | −0.1154 | 0.6628 | 0.5808 | 0.031* | |
| C2 | −0.214 (2) | 0.6785 (5) | 0.3162 (11) | 0.033 (2) | |
| H2 | −0.3380 | 0.6275 | 0.2804 | 0.040* | |
| C3 | 0.0498 (18) | 0.8069 (5) | 0.3117 (9) | 0.0188 (17) | |
| C4 | 0.2134 (18) | 0.8932 (5) | 0.2766 (9) | 0.0218 (17) | |
| C5 | 0.499 (2) | 1.0099 (5) | 0.3543 (10) | 0.0242 (17) | |
| H5 | 0.6276 | 1.0520 | 0.4205 | 0.029* | |
| C6 | 0.410 (2) | 1.0157 (6) | 0.1810 (11) | 0.034 (2) | |
| H6 | 0.4606 | 1.0630 | 0.1076 | 0.041* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Cu1 | 0.0243 (4) | 0.0286 (5) | 0.0165 (4) | −0.0008 (5) | −0.0027 (3) | −0.0001 (5) |
| Cl1 | 0.0319 (10) | 0.0354 (11) | 0.0210 (10) | 0.0008 (9) | −0.0032 (8) | 0.0051 (8) |
| Cl2 | 0.0275 (10) | 0.0375 (12) | 0.0241 (10) | −0.0049 (9) | −0.0047 (8) | −0.0030 (9) |
| N1 | 0.020 (3) | 0.026 (4) | 0.014 (3) | 0.000 (3) | −0.004 (2) | −0.002 (3) |
| N2 | 0.030 (4) | 0.036 (4) | 0.019 (3) | 0.000 (3) | −0.005 (3) | −0.002 (3) |
| N3 | 0.049 (4) | 0.029 (4) | 0.016 (3) | 0.005 (3) | −0.001 (3) | 0.004 (3) |
| N4 | 0.030 (4) | 0.024 (3) | 0.019 (3) | 0.002 (3) | −0.004 (3) | −0.002 (3) |
| C1 | 0.020 (4) | 0.035 (5) | 0.023 (4) | 0.004 (3) | 0.007 (3) | −0.005 (3) |
| C2 | 0.033 (5) | 0.023 (4) | 0.045 (5) | −0.008 (4) | 0.011 (4) | −0.014 (4) |
| C3 | 0.020 (4) | 0.030 (4) | 0.006 (3) | 0.008 (3) | −0.001 (3) | −0.008 (3) |
| C4 | 0.014 (4) | 0.031 (4) | 0.020 (4) | 0.009 (3) | 0.005 (3) | 0.007 (3) |
| C5 | 0.025 (4) | 0.020 (4) | 0.029 (4) | 0.000 (3) | 0.005 (3) | −0.009 (3) |
| C6 | 0.047 (5) | 0.030 (5) | 0.027 (5) | 0.005 (4) | 0.008 (4) | 0.001 (4) |
Geometric parameters (Å, º) top
| Cu1—N4 | 2.009 (6) | N3—C6 | 1.347 (10) |
| Cu1—N1 | 2.011 (6) | N3—H3A | 0.8600 |
| Cu1—Cl1 | 2.262 (2) | N4—C4 | 1.324 (9) |
| Cu1—Cl2 | 2.280 (2) | N4—C5 | 1.352 (10) |
| Cu1—Cl2i | 2.892 (2) | C1—C2 | 1.355 (11) |
| Cu1—Cu1i | 3.8671 (12) | C1—H1 | 0.9300 |
| N1—C3 | 1.339 (9) | C2—H2 | 0.9300 |
| N1—C1 | 1.374 (10) | C3—C4 | 1.464 (10) |
| N2—C3 | 1.314 (10) | C5—C6 | 1.348 (12) |
| N2—C2 | 1.370 (10) | C5—H5 | 0.9300 |
| N2—H2A | 0.8600 | C6—H6 | 0.9300 |
| N3—C4 | 1.308 (9) | | |
| | | |
| N4—Cu1—N1 | 80.8 (2) | C4—N4—Cu1 | 114.7 (5) |
| N4—Cu1—Cl1 | 169.71 (19) | C5—N4—Cu1 | 140.3 (5) |
| N1—Cu1—Cl1 | 91.79 (18) | C2—C1—N1 | 108.2 (7) |
| N4—Cu1—Cl2 | 89.83 (19) | C2—C1—H1 | 125.9 |
| N1—Cu1—Cl2 | 166.96 (18) | N1—C1—H1 | 125.9 |
| Cl1—Cu1—Cl2 | 96.27 (7) | C1—C2—N2 | 107.0 (7) |
| N4—Cu1—Cl2i | 95.03 (19) | C1—C2—H2 | 126.5 |
| N1—Cu1—Cl2i | 93.80 (17) | N2—C2—H2 | 126.5 |
| Cl1—Cu1—Cl2i | 92.58 (7) | N2—C3—N1 | 110.9 (7) |
| Cl2—Cu1—Cl2i | 96.07 (7) | N2—C3—C4 | 133.3 (7) |
| C3—N1—C1 | 106.1 (6) | N1—C3—C4 | 115.8 (7) |
| C3—N1—Cu1 | 113.4 (5) | N3—C4—N4 | 112.5 (7) |
| C1—N1—Cu1 | 139.9 (5) | N3—C4—C3 | 132.6 (7) |
| C3—N2—C2 | 107.8 (7) | N4—C4—C3 | 114.9 (6) |
| C3—N2—H2A | 126.1 | C6—C5—N4 | 108.6 (7) |
| C2—N2—H2A | 126.1 | C6—C5—H5 | 125.7 |
| C4—N3—C6 | 106.2 (7) | N4—C5—H5 | 125.7 |
| C4—N3—H3A | 126.9 | N3—C6—C5 | 107.7 (7) |
| C6—N3—H3A | 126.9 | N3—C6—H6 | 126.2 |
| C4—N4—C5 | 105.0 (6) | C5—C6—H6 | 126.2 |
| Symmetry code: (i) x−1, y, z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N3—H3A···Cl2ii | 0.86 | 2.44 | 3.221 (7) | 152 |
| N2—H2A···Cl1ii | 0.86 | 2.49 | 3.227 (7) | 145 |
| Symmetry code: (ii) x−1, y, z−1. |
Experimental details
| Crystal data |
| Chemical formula | [CuCl2(C6H6N4)] |
| Mr | 268.59 |
| Crystal system, space group | Monoclinic, P21 |
| Temperature (K) | 298 |
| a, b, c (Å) | 3.8671 (4), 14.8937 (17), 7.6363 (9) |
| β (°) | 93.742 (2) |
| V (Å3) | 438.88 (9) |
| Z | 2 |
| Radiation type | Mo Kα |
| µ (mm−1) | 3.05 |
| Crystal size (mm) | 0.34 × 0.06 × 0.05 |
| |
| Data collection |
| Diffractometer | Bruker APEX area-detector
diffractometer |
| Absorption correction | Multi-scan
(SADABS; Bruker, 2002) |
| Tmin, Tmax | 0.424, 0.863 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 4178, 1590, 1522 |
| Rint | 0.038 |
| (sin θ/λ)max (Å−1) | 0.600 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.053, 0.103, 1.19 |
| No. of reflections | 1590 |
| No. of parameters | 118 |
| No. of restraints | 1 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.70, −0.76 |
| Absolute structure | Flack (1983), with 761 Friedel pairs |
| Absolute structure parameter | 0.07 (3) |
Selected geometric parameters (Å, º) top| Cu1—N4 | 2.009 (6) | Cu1—Cl2 | 2.280 (2) |
| Cu1—N1 | 2.011 (6) | Cu1—Cl2i | 2.892 (2) |
| Cu1—Cl1 | 2.262 (2) | Cu1—Cu1i | 3.8671 (12) |
| | | |
| N4—Cu1—N1 | 80.8 (2) | Cl1—Cu1—Cl2 | 96.27 (7) |
| N4—Cu1—Cl1 | 169.71 (19) | N4—Cu1—Cl2i | 95.03 (19) |
| N1—Cu1—Cl1 | 91.79 (18) | N1—Cu1—Cl2i | 93.80 (17) |
| N4—Cu1—Cl2 | 89.83 (19) | Cl1—Cu1—Cl2i | 92.58 (7) |
| N1—Cu1—Cl2 | 166.96 (18) | Cl2—Cu1—Cl2i | 96.07 (7) |
| Symmetry code: (i) x−1, y, z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N3—H3A···Cl2ii | 0.86 | 2.44 | 3.221 (7) | 152 |
| N2—H2A···Cl1ii | 0.86 | 2.49 | 3.227 (7) | 145 |
| Symmetry code: (ii) x−1, y, z−1. |


 journal menu
journal menu metal-organic compounds
metal-organic compounds








![[kappa]](/logos/entities/kappa_rmgif.gif)
![[mu]](/logos/entities/mu_rmgif.gif) -chloro]
-chloro]


![[Figure 1]](http://journals.iucr.org/c/issues/2005/09/00/rb1010/rb1010fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2005/09/00/rb1010/rb1010fig2thm.gif)




2,2'-Biimidazole is a multifunctional ligand that can be coordinated to a transition metal in non-deprotonated (neutral, H2biim), mono-deprotonated (monoanion, Hbiim−) and di-deprotonated (dianion, biim2−) forms (Cancela et al., 2001). The metal-binding ability of its three forms has been well documented since the same 3d metal complexes with either neutral or deprotonated forms were first reported by Holmes et al. (1961). In the neutral state, H2biim possesses a double property, namely it can bind metals as either a bidentate chelate, seen, for example, with CuII (Liu & Su, 1996), VIV (Sang et al., 2002), and CoII and NiII (Atencio et al., 2004), or as a bridging ligand between two metal centres (Kirchner & Krebs, 1987), and can act as a donor in hydrogen-bonding interactions. Coordinated H2biim usually forms hydrogen bonds with counteranions and solvent molecules (Ye et al., 1999). On account of the bridging and counteranion properties of the Cl− anion, as well as the flexibility of the coordination stereochemistry of the CuII cation, we are interested in copper complexes with the Cl− anion and H2biim. In addition, copper(II) chloride yields adducts with H2biim in which the bi-heterocyclic ligand binds in the typical chelating mode, and one such complex, [Cu(H2biim)2]Cl2, has been reported previously (Atencio et al., 2005). The present study uses a similar method for synthesizing a new adduct with copper(II) chloride and H2biim by adjusting the molar ratio of copper(II) chloride and H2biim, which yielded the title compound, [Cu(C6H6N4)Cl2]n, (I).
In (I), each CuII ion is five-coordinated with a distorted square-pyramidal geometry (Fig. 1 and Table 1). The basal plane is formed by atoms N1 and N4 from one H2biim ligand in chelating mode, along with anions Cl1 and Cl2, with an r.m.s. deviation of 0.018 Å. The apical site is occupied by a Cl− anion, Cl2i [symmetry code: (i) x − 1, y, z], with a Cu—Cl bond distance of 2.892 (2) Å, which is longer than the average equatorial Cu—Cl bond distance of 2.271 (2) Å. The Cu—N bond distances and the chelating mode of the H2biim ligand are similar to those observed in [CuCl(C6H6N4)(H2O)][Cu(C4H5NO4Cl]·H2O (Gao et al., 2005). Atom Cu1 is located 0.1623 (8) Å out of the basal plane towards Cl2i.
Five-coordinate copper(II) complexes have geometries ranging from trigonal–bipyramidal to square-pyramidal. Energetically, the limiting trigonal–bipyramidal and square-pyramidal forms are often almost equally favourable, with a low activation barrier to interconversion. In the present instance, the observed geometry is very nearly square-pyramidal, as determined by the observed distortion value τ (van Albada et al., 1999; Addison et al., 1984) of 0.05, which is very near the ideal value of τ = 0. For perfect trigonal–bipyramidal geometry, τ = 1.
The bond distances and angles of the complexed H2biim ligand are unexceptional and compare well with those of the free ligand (Cromer et al., 1987). Of note is the dihedral angle between the two imidazole rings. The free ligand is found to crystallize in a trans conformation, with a dihedral angle of 4.6°, but in (I), the H2biim ligand takes on a cis conformation, with a dihedral angle of 2.3 (4)°. This angle is smaller than that observed in [Ag(NO3)(C6H6N4)]n [23.2 (2)°; Hester et al., 1997], in which H2biim functions as a bridging ligand to link the Ag atoms, forming a right-hand helical chain. In (I), the Cl2i anion connects adjacent [Cu(C6H6N4)Cl2] units to produce a linear chain along the crystallographic a axis, with a Cu1···Cu1i separation of 3.8671 (12) Å (Fig 1). In the chain, the heterocyclic rings are either parallel or nearly parallel to each other.
The above cis conformation and parallel disposition of H2biim preserve favourable π–π interactions between the two sets of parallel symmetry-related rings of the H2biim moieties [at (x, y, z) and (x − 1, y, z) or (x + 1, y, z)]. Ring centroids are separated by 3.867 (2) Å in both cases, and the perpendicular separations between the two sets of planes are 3.377 (3) and 3.351 (3) Å. These π–π interactions reinforce the linear chains. Adjacent linear chains are linked by N2—H2A···Cl1ii and N3—H3A···Cl2ii [symmetry code: (ii) x − 1, y, z − 1] hydrogen bonds along the [101] direction, resulting in a grid layer parallel to (010) (Fig. 2 and Table 2). The hydrogen-bonding pattern, as shown in Fig. 2, can be described in graph-set notation (Etter, 1990; Grell et al., 2000) as C22(9), R22(9), R22(14).