Ferric sulfate trihydrate has been synthesized at 403 K under hydrothermal conditions. The structure consists of quadruple chains of [Fe
2(SO
4)
3(H
2O)
3]
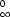
parallel to [010]. Each quadruple chain is composed of equal proportions of FeO
4(H
2O)
2 octahedra and FeO
5(H
2O) octahedra sharing corners with SO
4 tetrahedra. The chains are joined to each other by hydrogen bonds. This compound is a new hydration state of Fe
2(SO
4)
3·
nH
2O; minerals with
n = 0, 5, 7.25-7.75, 9 and 11 are found in nature.
Supporting information
α-Fe2O3 [1.000 (1) g] and sulfuric acid [1.939 (1) g] with a nominal
concentration of 95.9 wt% H2SO4 were mixed in a 23 ml Teflon-lined vessel,
sealed in a Parr stainless steel autoclave then stored in an isotemp oven set
at 403 K for 9 d. The product was an inhomogeneous, yellowish pink, solid
block. Examination with optical microscopy revealed transparent yellow
crystals embedded in a pink powdery matrix. The yellow crystals are cuboids or
short rectangular prisms, 100 to 300 µm on the longest edge and 40 to 100 µm
on the shortest edge. The structure of the yellow crystal was determined from
single-crystal X-ray diffraction to be the ferric sulfate trihydrate presented
above. The pink matrix was a combination of lausenite and rhomboclase
identified from powder X-ray diffraction (supporting material).
H atoms were found in difference Fourier maps and subsequently placed in
idealized positions with constrained distances of 0.90 Å (O—H), and with
Uiso(H) = 1.2Uiso of attached oxygen atoms.
Data collection: SMART (Bruker, 1998); cell refinement: SAINT-Plus (Bruker, 1998); data reduction: SAINT-Plus (Bruker, 1998); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008); molecular graphics: software?; software used to prepare material for publication: SHELXL97 (Sheldrick, 2008).
diiron(III) trisulfate trihydrate
top
Crystal data top
| Fe2(SO4)3·3H2O | F(000) = 904 |
| Mr = 453.93 | Dx = 2.656 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 855 reflections |
| a = 11.281 (3) Å | θ = 1.9–27.1° |
| b = 6.336 (2) Å | µ = 3.20 mm−1 |
| c = 16.278 (5) Å | T = 298 K |
| β = 102.676 (8)° | Block, yellow |
| V = 1135.1 (6) Å3 | 0.12 × 0.12 × 0.06 mm |
| Z = 4 | |
Data collection top
Bruker SMART CCD
diffractometer | 2309 independent reflections |
| Radiation source: fine-focus sealed tube | 1894 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.033 |
| ω and ϕ scans | θmax = 26.4°, θmin = 1.9° |
Absorption correction: multi-scan
(SADABS; Bruker, 2001) | h = −13→14 |
| Tmin = 0.700, Tmax = 0.831 | k = −7→7 |
| 7266 measured reflections | l = −20→20 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.028 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.084 | H-atom parameters constrained |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.045P)2 + 0.7177P]
where P = (Fo2 + 2Fc2)/3 |
| 2309 reflections | (Δ/σ)max < 0.001 |
| 199 parameters | Δρmax = 0.49 e Å−3 |
| 6 restraints | Δρmin = −0.55 e Å−3 |
Crystal data top
| Fe2(SO4)3·3H2O | V = 1135.1 (6) Å3 |
| Mr = 453.93 | Z = 4 |
| Monoclinic, P21/c | Mo Kα radiation |
| a = 11.281 (3) Å | µ = 3.20 mm−1 |
| b = 6.336 (2) Å | T = 298 K |
| c = 16.278 (5) Å | 0.12 × 0.12 × 0.06 mm |
| β = 102.676 (8)° | |
Data collection top
Bruker SMART CCD
diffractometer | 2309 independent reflections |
Absorption correction: multi-scan
(SADABS; Bruker, 2001) | 1894 reflections with I > 2σ(I) |
| Tmin = 0.700, Tmax = 0.831 | Rint = 0.033 |
| 7266 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.028 | 6 restraints |
| wR(F2) = 0.084 | H-atom parameters constrained |
| S = 1.05 | Δρmax = 0.49 e Å−3 |
| 2309 reflections | Δρmin = −0.55 e Å−3 |
| 199 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Fe1 | 0.02030 (3) | 0.25527 (5) | 0.11807 (2) | 0.01248 (13) | |
| Fe2 | 0.36121 (3) | 0.77262 (6) | 0.09097 (2) | 0.01340 (13) | |
| S1 | 0.07269 (6) | 0.75034 (9) | 0.09067 (4) | 0.01171 (16) | |
| S2 | 0.31145 (6) | 0.27291 (10) | 0.11913 (4) | 0.01403 (16) | |
| S3 | 0.26506 (6) | 0.72726 (10) | −0.11735 (4) | 0.01643 (17) | |
| O1 | 0.20241 (17) | 0.7803 (3) | 0.12936 (11) | 0.0172 (4) | |
| O2 | 0.05876 (18) | 0.6955 (3) | 0.00162 (11) | 0.0181 (4) | |
| O3 | 0.02430 (16) | 0.5769 (3) | 0.13491 (11) | 0.0171 (4) | |
| O4 | 0.00354 (17) | 0.9410 (3) | 0.10079 (12) | 0.0214 (4) | |
| O5 | 0.17839 (17) | 0.2632 (3) | 0.08359 (12) | 0.0185 (4) | |
| O6 | 0.36985 (17) | 0.0892 (3) | 0.08995 (12) | 0.0215 (4) | |
| O7 | 0.36109 (17) | 0.4588 (3) | 0.08306 (12) | 0.0239 (4) | |
| O8 | 0.33457 (19) | 0.2831 (3) | 0.21070 (12) | 0.0269 (5) | |
| O9 | 0.13156 (17) | 0.7477 (3) | −0.15455 (12) | 0.0180 (4) | |
| O10 | 0.28536 (19) | 0.7924 (3) | −0.02797 (12) | 0.0260 (5) | |
| O11 | 0.32929 (19) | 0.8742 (4) | −0.16031 (12) | 0.0338 (5) | |
| O12 | 0.2992 (2) | 0.5090 (4) | −0.12561 (15) | 0.0402 (6) | |
| O1W | 0.1057 (2) | 0.2204 (3) | 0.23977 (13) | 0.0253 (5) | |
| H1A | 0.182 (2) | 0.229 (5) | 0.257 (2) | 0.030* | |
| H1B | 0.072 (3) | 0.183 (5) | 0.2804 (17) | 0.030* | |
| O2W | 0.53224 (19) | 0.7845 (3) | 0.07628 (14) | 0.0236 (5) | |
| H2A | 0.585 (3) | 0.674 (4) | 0.0852 (19) | 0.028* | |
| H2B | 0.581 (3) | 0.892 (4) | 0.0890 (18) | 0.028* | |
| O3W | 0.4291 (2) | 0.7647 (3) | 0.21509 (13) | 0.0257 (5) | |
| H3A | 0.508 (2) | 0.761 (5) | 0.235 (2) | 0.031* | |
| H3B | 0.396 (3) | 0.733 (5) | 0.2572 (18) | 0.031* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Fe1 | 0.0121 (2) | 0.0114 (2) | 0.0142 (2) | −0.00009 (13) | 0.00330 (17) | −0.00003 (12) |
| Fe2 | 0.0127 (2) | 0.0114 (2) | 0.0160 (2) | −0.00012 (14) | 0.00287 (16) | 0.00014 (13) |
| S1 | 0.0114 (3) | 0.0098 (3) | 0.0142 (3) | −0.0001 (2) | 0.0033 (3) | 0.0002 (2) |
| S2 | 0.0119 (3) | 0.0113 (3) | 0.0190 (3) | −0.0005 (2) | 0.0035 (3) | −0.0002 (2) |
| S3 | 0.0123 (3) | 0.0205 (4) | 0.0167 (3) | 0.0007 (3) | 0.0038 (3) | −0.0019 (2) |
| O1 | 0.0134 (9) | 0.0209 (10) | 0.0174 (9) | −0.0009 (8) | 0.0036 (7) | −0.0011 (7) |
| O2 | 0.0173 (9) | 0.0209 (10) | 0.0155 (9) | −0.0034 (8) | 0.0024 (7) | 0.0002 (8) |
| O3 | 0.0214 (10) | 0.0116 (9) | 0.0200 (9) | −0.0013 (7) | 0.0082 (8) | −0.0003 (7) |
| O4 | 0.0183 (10) | 0.0114 (9) | 0.0364 (11) | 0.0002 (8) | 0.0101 (8) | −0.0017 (8) |
| O5 | 0.0119 (10) | 0.0232 (11) | 0.0205 (10) | 0.0005 (7) | 0.0036 (8) | 0.0006 (7) |
| O6 | 0.0212 (10) | 0.0105 (9) | 0.0351 (11) | 0.0012 (8) | 0.0113 (9) | 0.0005 (8) |
| O7 | 0.0248 (11) | 0.0122 (10) | 0.0382 (11) | −0.0012 (8) | 0.0148 (9) | 0.0005 (8) |
| O8 | 0.0203 (11) | 0.0410 (13) | 0.0186 (10) | −0.0012 (9) | 0.0022 (8) | −0.0025 (9) |
| O9 | 0.0134 (10) | 0.0238 (11) | 0.0169 (9) | 0.0017 (7) | 0.0038 (8) | 0.0006 (7) |
| O10 | 0.0208 (11) | 0.0385 (12) | 0.0179 (9) | −0.0020 (9) | 0.0025 (8) | −0.0020 (9) |
| O11 | 0.0291 (12) | 0.0477 (14) | 0.0266 (11) | −0.0160 (11) | 0.0106 (9) | −0.0016 (10) |
| O12 | 0.0353 (13) | 0.0293 (13) | 0.0518 (14) | 0.0146 (10) | 0.0008 (11) | −0.0092 (10) |
| O1W | 0.0177 (11) | 0.0403 (13) | 0.0172 (9) | 0.0001 (9) | 0.0026 (8) | 0.0063 (8) |
| O2W | 0.0147 (10) | 0.0199 (11) | 0.0380 (12) | 0.0022 (8) | 0.0101 (9) | 0.0028 (9) |
| O3W | 0.0174 (11) | 0.0421 (14) | 0.0164 (10) | −0.0014 (9) | 0.0013 (9) | 0.0031 (8) |
Geometric parameters (Å, º) top
| Fe1—O9i | 1.932 (2) | S2—O6 | 1.4668 (19) |
| Fe1—O2i | 1.9814 (19) | S2—O7 | 1.479 (2) |
| Fe1—O5 | 1.984 (2) | S2—O5 | 1.487 (2) |
| Fe1—O4ii | 2.0142 (19) | S3—O12 | 1.450 (2) |
| Fe1—O1W | 2.016 (2) | S3—O11 | 1.450 (2) |
| Fe1—O3 | 2.0553 (19) | S3—O10 | 1.481 (2) |
| Fe2—O10 | 1.941 (2) | S3—O9 | 1.500 (2) |
| Fe2—O7 | 1.993 (2) | O2—Fe1i | 1.9814 (19) |
| Fe2—O2W | 1.997 (2) | O4—Fe1iii | 2.0142 (19) |
| Fe2—O3W | 1.998 (2) | O6—Fe2ii | 2.008 (2) |
| Fe2—O6iii | 2.008 (2) | O9—Fe1i | 1.932 (2) |
| Fe2—O1 | 2.023 (2) | O1W—H1A | 0.85 (2) |
| S1—O2 | 1.4651 (19) | O1W—H1B | 0.87 (2) |
| S1—O4 | 1.4665 (19) | O2W—H2A | 0.91 (2) |
| S1—O1 | 1.473 (2) | O2W—H2B | 0.87 (2) |
| S1—O3 | 1.4821 (18) | O3W—H3A | 0.88 (2) |
| S2—O8 | 1.457 (2) | O3W—H3B | 0.88 (2) |
| | | |
| O9i—Fe1—O2i | 93.77 (8) | O2—S1—O3 | 109.51 (11) |
| O9i—Fe1—O5 | 178.31 (8) | O4—S1—O3 | 107.12 (11) |
| O2i—Fe1—O5 | 87.50 (8) | O1—S1—O3 | 108.97 (11) |
| O9i—Fe1—O4ii | 88.33 (7) | O8—S2—O6 | 111.98 (12) |
| O2i—Fe1—O4ii | 90.58 (8) | O8—S2—O7 | 111.85 (12) |
| O5—Fe1—O4ii | 92.76 (7) | O6—S2—O7 | 105.49 (12) |
| O9i—Fe1—O1W | 87.80 (9) | O8—S2—O5 | 109.79 (12) |
| O2i—Fe1—O1W | 176.82 (8) | O6—S2—O5 | 109.15 (11) |
| O5—Fe1—O1W | 90.88 (9) | O7—S2—O5 | 108.44 (11) |
| O4ii—Fe1—O1W | 92.23 (8) | O12—S3—O11 | 113.38 (14) |
| O9i—Fe1—O3 | 87.92 (7) | O12—S3—O10 | 111.72 (13) |
| O2i—Fe1—O3 | 88.11 (7) | O11—S3—O10 | 107.99 (12) |
| O5—Fe1—O3 | 91.02 (7) | O12—S3—O9 | 107.97 (12) |
| O4ii—Fe1—O3 | 175.94 (8) | O11—S3—O9 | 108.26 (12) |
| O1W—Fe1—O3 | 89.18 (8) | O10—S3—O9 | 107.31 (12) |
| O10—Fe2—O7 | 90.35 (8) | S1—O1—Fe2 | 136.79 (12) |
| O10—Fe2—O2W | 95.98 (9) | S1—O2—Fe1i | 149.64 (13) |
| O7—Fe2—O2W | 90.96 (8) | S1—O3—Fe1 | 132.06 (11) |
| O10—Fe2—O3W | 175.78 (9) | S1—O4—Fe1iii | 142.56 (12) |
| O7—Fe2—O3W | 92.01 (8) | S2—O5—Fe1 | 141.66 (13) |
| O2W—Fe2—O3W | 87.49 (9) | S2—O6—Fe2ii | 139.79 (12) |
| O10—Fe2—O6iii | 86.49 (8) | S2—O7—Fe2 | 139.77 (12) |
| O7—Fe2—O6iii | 174.52 (8) | S3—O9—Fe1i | 138.94 (12) |
| O2W—Fe2—O6iii | 84.93 (8) | S3—O10—Fe2 | 154.28 (15) |
| O3W—Fe2—O6iii | 91.42 (8) | Fe1—O1W—H1A | 123 (2) |
| O10—Fe2—O1 | 94.59 (9) | Fe1—O1W—H1B | 126 (2) |
| O7—Fe2—O1 | 93.19 (7) | H1A—O1W—H1B | 111 (3) |
| O2W—Fe2—O1 | 168.62 (8) | Fe2—O2W—H2A | 125 (2) |
| O3W—Fe2—O1 | 81.78 (9) | Fe2—O2W—H2B | 125 (2) |
| O6iii—Fe2—O1 | 91.52 (7) | H2A—O2W—H2B | 102 (3) |
| O2—S1—O4 | 111.23 (11) | Fe2—O3W—H3A | 121 (2) |
| O2—S1—O1 | 109.53 (11) | Fe2—O3W—H3B | 131 (2) |
| O4—S1—O1 | 110.43 (11) | H3A—O3W—H3B | 106 (3) |
| Symmetry codes: (i) −x, −y+1, −z; (ii) x, y−1, z; (iii) x, y+1, z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1W—H1A···O8 | 0.85 (2) | 2.05 (3) | 2.754 (3) | 140 (3) |
| O1W—H1B···O3iv | 0.87 (2) | 2.05 (2) | 2.907 (3) | 173 (3) |
| O2W—H2A···O12v | 0.91 (2) | 1.76 (2) | 2.655 (3) | 166 (3) |
| O2W—H2B···O11vi | 0.87 (2) | 2.01 (2) | 2.837 (3) | 157 (3) |
| O3W—H3A···O8vii | 0.88 (2) | 1.81 (2) | 2.677 (3) | 171 (3) |
| O3W—H3B···O11viii | 0.88 (2) | 1.81 (2) | 2.676 (3) | 171 (3) |
| Symmetry codes: (iv) −x, y−1/2, −z+1/2; (v) −x+1, −y+1, −z; (vi) −x+1, −y+2, −z; (vii) −x+1, y+1/2, −z+1/2; (viii) x, −y+3/2, z+1/2. |
Experimental details
| Crystal data |
| Chemical formula | Fe2(SO4)3·3H2O |
| Mr | 453.93 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 298 |
| a, b, c (Å) | 11.281 (3), 6.336 (2), 16.278 (5) |
| β (°) | 102.676 (8) |
| V (Å3) | 1135.1 (6) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 3.20 |
| Crystal size (mm) | 0.12 × 0.12 × 0.06 |
| |
| Data collection |
| Diffractometer | Bruker SMART CCD
diffractometer |
| Absorption correction | Multi-scan
(SADABS; Bruker, 2001) |
| Tmin, Tmax | 0.700, 0.831 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 7266, 2309, 1894 |
| Rint | 0.033 |
| (sin θ/λ)max (Å−1) | 0.625 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.028, 0.084, 1.05 |
| No. of reflections | 2309 |
| No. of parameters | 199 |
| No. of restraints | 6 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.49, −0.55 |
Selected geometric parameters (Å, º) top| Fe1—O9i | 1.932 (2) | Fe2—O10 | 1.941 (2) |
| Fe1—O2i | 1.9814 (19) | Fe2—O7 | 1.993 (2) |
| Fe1—O5 | 1.984 (2) | Fe2—O2W | 1.997 (2) |
| Fe1—O4ii | 2.0142 (19) | Fe2—O3W | 1.998 (2) |
| Fe1—O1W | 2.016 (2) | Fe2—O6iii | 2.008 (2) |
| Fe1—O3 | 2.0553 (19) | Fe2—O1 | 2.023 (2) |
| | | |
| O9i—Fe1—O2i | 93.77 (8) | O5—Fe1—O1W | 90.88 (9) |
| O9i—Fe1—O5 | 178.31 (8) | O4ii—Fe1—O1W | 92.23 (8) |
| O2i—Fe1—O5 | 87.50 (8) | O9i—Fe1—O3 | 87.92 (7) |
| O9i—Fe1—O4ii | 88.33 (7) | O2i—Fe1—O3 | 88.11 (7) |
| O2i—Fe1—O4ii | 90.58 (8) | O5—Fe1—O3 | 91.02 (7) |
| O5—Fe1—O4ii | 92.76 (7) | O4ii—Fe1—O3 | 175.94 (8) |
| O9i—Fe1—O1W | 87.80 (9) | O1W—Fe1—O3 | 89.18 (8) |
| O2i—Fe1—O1W | 176.82 (8) | | |
| Symmetry codes: (i) −x, −y+1, −z; (ii) x, y−1, z; (iii) x, y+1, z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1W—H1A···O8 | 0.85 (2) | 2.05 (3) | 2.754 (3) | 140 (3) |
| O1W—H1B···O3iv | 0.87 (2) | 2.05 (2) | 2.907 (3) | 173 (3) |
| O2W—H2A···O12v | 0.91 (2) | 1.76 (2) | 2.655 (3) | 166 (3) |
| O2W—H2B···O11vi | 0.87 (2) | 2.01 (2) | 2.837 (3) | 157 (3) |
| O3W—H3A···O8vii | 0.88 (2) | 1.81 (2) | 2.677 (3) | 171 (3) |
| O3W—H3B···O11viii | 0.88 (2) | 1.81 (2) | 2.676 (3) | 171 (3) |
| Symmetry codes: (iv) −x, y−1/2, −z+1/2; (v) −x+1, −y+1, −z; (vi) −x+1, −y+2, −z; (vii) −x+1, y+1/2, −z+1/2; (viii) x, −y+3/2, z+1/2. |


 journal menu
journal menu inorganic compounds
inorganic compounds












![[Figure 1]](http://journals.iucr.org/c/issues/2011/05/00/qs3002/qs3002fig1thm.gif)




Ferric sulfate trihydrate belongs to a group of ferric sulfates sharing a general formula Fe2(SO4)3.nH2O, that includes mikasaite (n = 0) (Miura et al., 1994), lausenite (n = 5) (Majzlan et al., 2005), kornelite (n = 7.25–7.75) (Ackermann et al., 2009; Robinson & Fang, 1973), paracoquimbite (n = 9) (Robinson & Fang, 1971), quenstedtite (n = 11) (Thomas et al., 1974) and a synthetic Fe2(SO4)3 (n = 0), a polymorph of mikasaite (Christidis & Rentzeperis, 1975). These ferric sulfate minerals are usually found as efflorescence in acid mine drainage (AMD) areas, formed as oxidation products of pyrite (FeS2) and other sulfide minerals (Jambor et al., 2000). Other commonly found ferric sulfates in AMD regions include coquimbite [(Fe,Al)2(SO4)3.9H2O], jarosite [KFe3(SO4)2(OH)], rhomboclase [(H5O2)Fe(SO4)2.2H2O] and copiapite [Fe2+Fe3+4(SO4)2(OH)2.20H2O]. Dissolution of these minerals greatly increases water acidity because of ferric ion hydrolysis. Plus, these ferric sulfates usually absorb or co-precipitate with toxic metals such as Cr and Pb, and act as a secondary source of these toxic metal pollutants in local waters as they dissolve (Jambor et al., 2000). To better control and reduce the adverse environmental effects of ferric sulfates require a thorough understanding of the behavior and stability of these minerals as functions of environmental factors, such as pH, relative humidity and temperature. Understanding the transformations of the ferric sulfates as a function of environmental conditions has been the focus of recent studies (Ackermann et al., 2009; Majzlan, 2010; Tosca et al., 2007; Xu et al., 2009, 2010).
One difficulty in delineating phase stability relationships in the FeIII–SO4–H2O system is the need for correct and complete crystal structure models for all phases in the system. For example, paracoquimbite and coquimbite had for a long time been considered as polymorphs (Fang & Robinson, 1970) until a recent study showing the amount of aluminium in the mineral determines the structure type: paracoquimbite is pure Fe2(SO4)3.9H2O, while coquimbite has an Al/Fe ratio close to 1/3 with Al predominantly occupying the [M(H2O)6]3+ metal site (Majzlan et al., 2010). A third structure type in this coquimbite series was recently found in a mineral with a 1:1 Al/Fe ratio (Demartin et al., 2010). Further, synthesis of ferric sulfates often produces new phases (Chipera et al., 2007; Freeman et al., 2009; Majzlan et al., 2005; Peterson et al., 2009; Xu et al., 2009). Structures of these new phases are mostly unresolved because of their occurrence only as fine-grained mixed-phase powders. During the course of our survey of the FeIII–SO4–H2O system we discovered a new phase, ferric sulfate trihydrate.
The structure of trihydrate contains identical quadruple chains of [Fe2(SO4)3(H2O)3]∞0 parallel to [010], as shown in Fig. 1. Each quadruple chain consists of four single chains of alternating FeO6 octahedra and SO4 tetrahedra extending along the b axis. Of the four single chains, two are symmetrically independent as `–Fe(1)—S(1)—Fe(1)–' and `–Fe(2)—S(2)—Fe(2)–'; the other two are generated through an inversion center. A third sulfate group S(3)O4 connects to Fe(1)O6 and Fe(2)O6 by corner sharing. Of the three unique water molecules, one (O1W) is coordinated to the Fe(1) site, and the other two (O2W and O3W) are coordinated to the Fe(2) site.
The quadruple chains are linked to one another by water–sulfate O—H···O hydrogen bonds (Table 2). All hydrogen bonds involve terminal O in SO4 groups as the acceptor, except the O1W—H1B···O3ii [symmetry code: (ii) -x, y - 1/2, -z + 1/2] hydrogen bond, where the acceptor O bridges Fe(1) and S(1). There is also one intramolecular hydrogen bond, O1W—H1B···O8.
The quadruple chain structure of the ferric sulfate trihydrate has no similar counterparts in any known sulfates. Though unique, the trihydrate structure shares similarities to other phases in the Fe2(SO4)3.nH2O group. For example, the FeO6 octahedra do not directly connect with each other but are linked via corner-sharing SO4 tetrahedra. It should be mentioned that FeO6 octahedra do share corners in some other ferric sulfates such as copiapite and jarosite.
Including the trihydrate described here, there are a total of six hydration states of Fe2(SO4)3.nH2O; namely n = 0, 3, 5, 7.25–7.75, 9 and 11. Anhydrous ferric sulfate has two polymorphs, a monoclinic form and a trigonal form. The trigonal form occurs in nature as the mineral mikasaite, while the synthetic monoclinic form has no mineral equivalent (Christidis & Rentzeperis, 1975; Miura et al., 1994). Both forms consist of frameworks of connected FeO6 octahedra and SO4 tetrahedra. The structure of lausenite, Fe2(SO4)3.5H2O, is composed of corrugated slabs of [Fe2(SO4)3(H2O)5]∞0 (Majzlan et al., 2005). The five water molecules shown in the formula are all coordinated to the two Fe sites, two coordinated to one and three coordinated to the other. The structure of kornelite, Fe2(SO4)3.7.25–7.75H2O, consists of slabs similar to the ones in lausenite, with a formula [Fe2(SO4)3(H2O)6]∞0. Each of the two Fe sites is bonded to three water molecules. The remaining 1.25 to 1.75 water molecules shown in the formula are located between neighboring slabs as isolated water (Robinson & Fang, 1973). The paracoquimbite structure contains isolated clusters of [Fe(H2O)6]3+ and [Fe3(SO4)6(H2O)6]3- and six uncoordinated water molecules (Robinson & Fang, 1971). The structure of quenstedtite, Fe2(SO4)3.11H2O, consists of isolated clusters of [Fe(SO4)(H2O)5]+ and [Fe(SO4)2(H2O)4]- and two uncoordinated water molecules (Thomas et al., 1974).
As the hydration state increases, FeO6 octahedra and SO4 tetrahedra tend to be disassociated, as more water molecules coordinate to Fe to form simple clusters. The hydration state of trihydrate lies in between that of anhydrous ferric sulfate and lausenite. Like lausenite, the structure of the trihydrate does not have uncoordinated water molecules as found in kornelite and higher hydration states. As opposed to limited clusters in the higher hyration states, both the trihydrate and the lausenite have infinite clusters, one-dimensional chains for the trihydrate and two-dimensional slabs for the lausenite. Further, the quadruple chain of trihydrate has three terminal O sites for water molecules, while the slab of lausenite has five. This appears to be counterintuitive: a common first impression would be that a chain arrangement has more terminal O sites than two-dimensional sheets. It is easy to understand this if one considers the fact that a quadruple chain has much less [far fewer] terminal sites than four single chains; also, the slab in lausenite is far from [less like] a dense sheet such as the MnO2 layer in birnessite, [and closer to] but closer to a net of interlaced chains.
Therefore, the trihydrate, though having a unique quadruple chain structure, shares the basic structural features present in other phases in the Fe2(SO4)3.nH2O system. The trihydrate structure also fits in the trend of structural changes set by hydration levels.