

 journal menu
journal menu research papers
research papers









The crystal structure of the red polymorph of tetrahexylsexithiophene (THST) is solved from X-ray powder diffraction data by a direct-space Monte Carlo simulated-annealing approach. First-principles density functional theory (DFT) calculations are used to distinguish between three nearly identical solutions in the space groups C2/m, C2 and P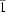 and to improve the overall accuracy of the crystal structure. The correct space group is found to be C2/m. In all space groups, the thiophene backbone is planar and the hexyl side chains assume an all-trans conformation except for two terminal methyl residues, which adopt a gauche orientation. The ability of first-principles DFT calculations to provide atomic coordinates of single-crystal quality is demonstrated by lattice-energy minimization of the known crystal structure of the yellow polymorph of THST. The combination of Monte Carlo simulated annealing, first-principles DFT calculations and Rietveld refinement presented in this paper is generally applicable. It provides a powerful alternative to standard approaches in cases where the information content of the powder diffraction pattern alone is insufficient to distinguish between different structure solutions. DFT calculations can also provide invaluable guidance in Rietveld refinement.
and to improve the overall accuracy of the crystal structure. The correct space group is found to be C2/m. In all space groups, the thiophene backbone is planar and the hexyl side chains assume an all-trans conformation except for two terminal methyl residues, which adopt a gauche orientation. The ability of first-principles DFT calculations to provide atomic coordinates of single-crystal quality is demonstrated by lattice-energy minimization of the known crystal structure of the yellow polymorph of THST. The combination of Monte Carlo simulated annealing, first-principles DFT calculations and Rietveld refinement presented in this paper is generally applicable. It provides a powerful alternative to standard approaches in cases where the information content of the powder diffraction pattern alone is insufficient to distinguish between different structure solutions. DFT calculations can also provide invaluable guidance in Rietveld refinement.
Keywords: direct-space Monte Carlo simulated annealing; density functional theory; lattice-energy minimization; Rietveld refinement; powder diffraction; tetrahexylsexithiophene.
Supporting information
 | Crystallographic Information File (CIF) https://doi.org/10.1107/S0021889802002844/ks0106sup1.cif |
CCDC reference: 214226
Computing details top
![[Figure 1]](http://journals.iucr.org/j/issues/2002/03/00/ks0106/ks0106fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/j/issues/2002/03/00/ks0106/ks0106fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/j/issues/2002/03/00/ks0106/ks0106fig3thm.gif)
(C2m_best_0k03shift) top
Crystal data top
| ? | β = 117.2444° |
| Mr = ? | V = ? Å3 |
| Monoclinic, C2/M | Z = ? |
| a = 24.9512 Å | ? radiation, λ = ? Å |
| b = 7.5153 Å | × × mm |
| c = 13.8689 Å |
Crystal data top
| ? | β = 117.2444° |
| Mr = ? | V = ? Å3 |
| Monoclinic, C2/M | Z = ? |
| a = 24.9512 Å | ? radiation, λ = ? Å |
| b = 7.5153 Å | × × mm |
| c = 13.8689 Å |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top
| x | y | z | Uiso*/Ueq | ||
| C1 | −0.08856 | 0.00691 | 0.92701 | 0.00000* | |
| H2 | −0.16003 | −0.11231 | 0.78473 | 0.00000* | |
| C3 | −0.04600 | −0.00447 | 0.57910 | 0.00000* | |
| C4 | −0.46479 | 0.01074 | 0.30247 | 0.00000* | |
| C5 | −0.10866 | −0.00395 | 0.54396 | 0.00000* | |
| H6 | −0.06622 | −0.12427 | 0.76129 | 0.00000* | |
| S7 | −0.25669 | 0.00000 | 0.41614 | 0.00000* | |
| H8 | −0.15741 | −0.06355 | 0.97291 | 0.00000* | |
| H9 | −0.16716 | −0.12080 | 0.59950 | 0.00000* | |
| H10 | −0.42353 | −0.11068 | 0.52283 | 0.00000* | |
| H11 | −0.36131 | −0.06277 | 0.90081 | 0.00000* | |
| C12 | −0.36105 | 0.00265 | 0.47202 | 0.00000* | |
| H13 | −0.06448 | 0.11083 | 0.76004 | 0.00000* | |
| H14 | −0.05971 | −0.08791 | 1.14326 | 0.00000* | |
| H15 | −0.31567 | 0.23772 | 0.90801 | 0.00000* | |
| H16 | −0.53947 | 0.01438 | 0.13524 | 0.00000* | |
| S17 | −0.43974 | 0.00516 | 0.14313 | 0.00000* | |
| C18 | −0.31917 | 0.00000 | 0.28804 | 0.00000* | |
| C19 | −0.13721 | −0.00459 | 0.61819 | 0.00000* | |
| H20 | −0.21500 | −0.00686 | 0.19846 | 0.00000* | |
| H21 | −0.33096 | −0.00133 | 0.12283 | 0.00000* | |
| H22 | −0.10666 | 0.07388 | 1.15927 | 0.00000* | |
| H23 | −0.31667 | 0.10894 | 0.68397 | 0.00000* | |
| H24 | −0.26170 | 0.08578 | 0.99793 | 0.00000* | |
| C25 | −0.39276 | 0.00364 | 0.54340 | 0.00000* | |
| H26 | −0.06327 | −0.11865 | 0.95249 | 0.00000* | |
| H27 | −0.33186 | −0.11566 | 0.49303 | 0.00000* | |
| C28 | −0.29985 | −0.00207 | 0.20894 | 0.00000* | |
| C29 | −0.49193 | 0.01123 | 0.19156 | 0.00000* | |
| C30 | −0.37771 | −0.00869 | 0.73744 | 0.00000* | |
| C31 | −0.12943 | 0.00265 | 0.80522 | 0.00000* | |
| S32 | −0.09361 | −0.00323 | 0.37108 | 0.00000* | |
| C33 | −0.02942 | −0.00398 | 0.49672 | 0.00000* | |
| C34 | −0.14180 | −0.00366 | 0.43097 | 0.00000* | |
| C35 | −0.20551 | −0.00292 | 0.36261 | 0.00000* | |
| C36 | −0.23707 | −0.00388 | 0.25000 | 0.00000* | |
| C37 | −0.37965 | 0.00307 | 0.27467 | 0.00000* | |
| C38 | −0.40096 | 0.00589 | 0.35165 | 0.00000* | |
| C39 | −0.09467 | −0.00472 | 0.73930 | 0.00000* | |
| C40 | −0.12244 | 0.03888 | 0.99279 | 0.00000* | |
| C41 | −0.08155 | 0.04152 | 1.11442 | 0.00000* | |
| C42 | −0.34756 | −0.00501 | 0.66359 | 0.00000* | |
| C43 | −0.33464 | −0.03835 | 0.85744 | 0.00000* | |
| C44 | −0.29114 | 0.11446 | 0.91212 | 0.00000* | |
| H45 | −0.01305 | −0.00606 | 0.66410 | 0.00000* | |
| H46 | −0.49014 | 0.01403 | 0.34822 | 0.00000* | |
| H47 | −0.16703 | 0.11186 | 0.60005 | 0.00000* | |
| H48 | −0.15834 | 0.12158 | 0.78150 | 0.00000* | |
| H49 | −0.05469 | 0.11229 | 0.94557 | 0.00000* | |
| H50 | −0.14677 | 0.16594 | 0.96765 | 0.00000* | |
| H51 | −0.04574 | 0.14102 | 1.13603 | 0.00000* | |
| H52 | −0.33003 | 0.11687 | 0.49534 | 0.00000* | |
| H53 | −0.42031 | 0.12447 | 0.52751 | 0.00000* | |
| H54 | −0.31965 | −0.12508 | 0.67840 | 0.00000* | |
| H55 | −0.40255 | 0.11646 | 0.72770 | 0.00000* | |
| H56 | −0.41145 | −0.11568 | 0.70980 | 0.00000* | |
| H57 | −0.30896 | −0.16127 | 0.86614 | 0.00000* | |
| H58 | −0.26201 | 0.13990 | 0.87403 | 0.00000* |
Bond lengths (Å) top
| C1—C31 | 1.522 | C18—C28 | 1.386 |
| C1—C40 | 1.520 | C18—C37 | 1.434 |
| C1—H49 | 1.099 | C19—H47 | 1.101 |
| C1—H26 | 1.100 | C19—C39 | 1.522 |
| H2—C31 | 1.101 | H20—C36 | 1.084 |
| C3—C5 | 1.409 | H21—C28 | 1.086 |
| C3—C33 | 1.382 | H22—C41 | 1.093 |
| C3—H45 | 1.086 | H23—C42 | 1.099 |
| C4—H46 | 1.082 | H24—C44 | 1.096 |
| C4—C29 | 1.368 | C25—H53 | 1.099 |
| C4—C38 | 1.417 | C25—C42 | 1.528 |
| C5—C19 | 1.496 | C28—C36 | 1.401 |
| C5—C34 | 1.397 | C30—C43 | 1.529 |
| H6—C39 | 1.098 | C30—C42 | 1.524 |
| S7—C35 | 1.747 | C30—H55 | 1.100 |
| S7—C18 | 1.747 | C30—H56 | 1.099 |
| H8—C40 | 1.100 | C31—H48 | 1.100 |
| H9—C19 | 1.100 | C31—C39 | 1.522 |
| H10—C25 | 1.099 | S32—C34 | 1.746 |
| H11—C43 | 1.098 | S32—C33 | 1.745 |
| C12—C38 | 1.502 | C34—C35 | 1.430 |
| C12—H27 | 1.101 | C35—C36 | 1.390 |
| C12—H52 | 1.101 | C37—C38 | 1.394 |
| C12—C25 | 1.524 | C40—H50 | 1.099 |
| H13—C39 | 1.098 | C40—C41 | 1.520 |
| H14—C41 | 1.097 | C41—H51 | 1.096 |
| H15—C44 | 1.097 | C42—H54 | 1.100 |
| H16—C29 | 1.081 | C43—H57 | 1.099 |
| S17—C37 | 1.753 | C43—C44 | 1.523 |
| S17—C29 | 1.717 | C44—H58 | 1.093 |
Experimental details
| Crystal data | |
| Chemical formula | ? |
| Mr | ? |
| Crystal system, space group | Monoclinic, C2/M |
| Temperature (K) | ? |
| a, b, c (Å) | 24.9512, 7.5153, 13.8689 |
| β (°) | 117.2444 |
| V (Å3) | ? |
| Z | ? |
| Radiation type | ?, λ = ? Å |
| µ (mm−1) | ? |
| Crystal size (mm) | × × |
| Data collection | |
| Diffractometer | ? |
| Absorption correction | ? |
| No. of measured, independent and observed (?) reflections | ?, ?, ? |
| Rint | ? |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | ?, ?, ? |
| No. of reflections | ? |
| No. of parameters | ? |
| No. of restraints | ? |
| Δρmax, Δρmin (e Å−3) | ?, ? |



