In the title compound, C
20H
16Cl
2O
4S
2, the molecules lie across centres of inversion. A single type of intermolecular C—H

O hydrogen bond, with a C
O distance of 3.254 (3) Å and a C—H
O angle of 132°, links the molecules into ladders whose uprights form
C(6) chains and whose rungs enclose centrosymmetric
R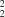
(22) rings.
Supporting information
CCDC reference: 147658
A sample of 2,5-bis(4-chlorophenylthio)-1,4-dimethylbenzene (Grant et al., 1987) was oxidized using sodium periodate in CH2Cl2 to yield (I). Crystals suitable for single-crystal X-ray diffraction were grown from ethanol solution (m.p. 506–508 K).
Compound (I) crystallized in the triclinic system; space group P1 was assumed and confirmed by the analysis. H atoms were treated as riding, with C—H 0.95 (aromatic) and 0.98 Å (methyl). Examination of the structure with PLATON (Spek, 1999) showed that there were no solvent accessible voids in the crystal lattice.
Data collection: KappaCCD Server Software (Nonius, 1997); cell refinement: DENZO (Otwinowski & Minor, 1997); data reduction: DENZO; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: PLATON (Spek, 1999); software used to prepare material for publication: SHELXL97 and PRPKAPPA (Ferguson, 1999).
1,4-bis(4-chlorophenylsulfonyl)-2,5-dimethylbenzene
top
Crystal data top
| C20H16Cl2O4S2 | Z = 1 |
| Mr = 455.35 | F(000) = 234 |
| Triclinic, P1 | Dx = 1.605 Mg m−3 |
| a = 5.4859 (2) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 6.9595 (4) Å | Cell parameters from 2112 reflections |
| c = 12.5211 (7) Å | θ = 1.7–27.5° |
| α = 98.6245 (17)° | µ = 0.59 mm−1 |
| β = 93.561 (4)° | T = 150 K |
| γ = 92.174 (3)° | Plate, colourless |
| V = 471.18 (4) Å3 | 0.10 × 0.10 × 0.03 mm |
Data collection top
KappaCCD
diffractometer | 2112 independent reflections |
| Radiation source: fine-focus sealed X-ray tube | 1689 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.039 |
| ϕ scans and ω scans with κ offsets | θmax = 27.5°, θmin = 1.7° |
Absorption correction: multi-scan
(SORTAV; Blessing, 1995, 1997) | h = −7→7 |
| Tmin = 0.943, Tmax = 0.985 | k = −9→8 |
| 5965 measured reflections | l = −16→16 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.039 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.104 | H-atom parameters constrained |
| S = 1.07 | Calculated w = 1/[σ2(Fo2) + (0.0411P)2 + 0.1338P]
where P = (Fo2 + 2Fc2)/3 |
| 2112 reflections | (Δ/σ)max < 0.001 |
| 128 parameters | Δρmax = 0.39 e Å−3 |
| 0 restraints | Δρmin = −0.53 e Å−3 |
Crystal data top
| C20H16Cl2O4S2 | γ = 92.174 (3)° |
| Mr = 455.35 | V = 471.18 (4) Å3 |
| Triclinic, P1 | Z = 1 |
| a = 5.4859 (2) Å | Mo Kα radiation |
| b = 6.9595 (4) Å | µ = 0.59 mm−1 |
| c = 12.5211 (7) Å | T = 150 K |
| α = 98.6245 (17)° | 0.10 × 0.10 × 0.03 mm |
| β = 93.561 (4)° | |
Data collection top
KappaCCD
diffractometer | 2112 independent reflections |
Absorption correction: multi-scan
(SORTAV; Blessing, 1995, 1997) | 1689 reflections with I > 2σ(I) |
| Tmin = 0.943, Tmax = 0.985 | Rint = 0.039 |
| 5965 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.039 | 0 restraints |
| wR(F2) = 0.104 | H-atom parameters constrained |
| S = 1.07 | Δρmax = 0.39 e Å−3 |
| 2112 reflections | Δρmin = −0.53 e Å−3 |
| 128 parameters | |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| S1 | 0.85567 (9) | 0.33050 (7) | 0.32509 (4) | 0.01934 (17) | |
| Cl1 | 0.20835 (10) | −0.31579 (8) | 0.02434 (5) | 0.03541 (19) | |
| O1 | 1.0591 (2) | 0.2411 (2) | 0.37152 (12) | 0.0244 (3) | |
| O2 | 0.9031 (3) | 0.4838 (2) | 0.26234 (12) | 0.0252 (3) | |
| C1 | 0.3882 (4) | −0.1363 (3) | 0.10821 (16) | 0.0227 (5) | |
| C2 | 0.6042 (4) | −0.1866 (3) | 0.15770 (16) | 0.0235 (5) | |
| H2 | 0.6548 | −0.3166 | 0.1451 | 0.028* | |
| C3 | 0.7455 (4) | −0.0436 (3) | 0.22619 (17) | 0.0218 (4) | |
| H3 | 0.8928 | −0.0755 | 0.2621 | 0.026* | |
| C4 | 0.6701 (4) | 0.1460 (3) | 0.24174 (16) | 0.0185 (4) | |
| C5 | 0.4549 (4) | 0.1954 (3) | 0.19024 (17) | 0.0225 (5) | |
| H5 | 0.4062 | 0.3261 | 0.2012 | 0.027* | |
| C6 | 0.3121 (4) | 0.0531 (3) | 0.12291 (17) | 0.0246 (5) | |
| H6 | 0.1642 | 0.0845 | 0.0873 | 0.030* | |
| C7 | 0.6607 (3) | 0.4218 (3) | 0.42770 (16) | 0.0177 (4) | |
| C8 | 0.5380 (3) | 0.5875 (3) | 0.41112 (16) | 0.0194 (4) | |
| H8 | 0.5674 | 0.6454 | 0.3488 | 0.023* | |
| C9 | 0.3745 (3) | 0.6708 (3) | 0.48236 (16) | 0.0182 (4) | |
| C91 | 0.2438 (4) | 0.8469 (3) | 0.45704 (18) | 0.0244 (5) | |
| H91A | 0.0696 | 0.8119 | 0.4399 | 0.037* | |
| H91B | 0.3134 | 0.8934 | 0.3948 | 0.037* | |
| H91C | 0.2635 | 0.9499 | 0.5200 | 0.037* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| S1 | 0.0173 (3) | 0.0193 (3) | 0.0204 (3) | 0.00115 (19) | 0.0020 (2) | −0.0005 (2) |
| Cl1 | 0.0337 (3) | 0.0341 (3) | 0.0332 (4) | −0.0099 (2) | −0.0024 (3) | −0.0071 (2) |
| O1 | 0.0165 (7) | 0.0274 (8) | 0.0276 (8) | 0.0048 (6) | −0.0008 (6) | −0.0008 (6) |
| O2 | 0.0258 (8) | 0.0235 (7) | 0.0270 (8) | −0.0001 (6) | 0.0069 (7) | 0.0044 (6) |
| C1 | 0.0248 (11) | 0.0245 (11) | 0.0168 (10) | −0.0068 (9) | 0.0028 (9) | −0.0021 (8) |
| C2 | 0.0301 (12) | 0.0198 (10) | 0.0205 (11) | 0.0023 (8) | 0.0043 (9) | 0.0013 (8) |
| C3 | 0.0222 (11) | 0.0226 (10) | 0.0207 (11) | 0.0031 (8) | 0.0012 (9) | 0.0031 (8) |
| C4 | 0.0189 (10) | 0.0202 (10) | 0.0156 (10) | −0.0001 (8) | 0.0026 (8) | 0.0003 (8) |
| C5 | 0.0215 (11) | 0.0224 (10) | 0.0235 (11) | 0.0043 (8) | 0.0023 (9) | 0.0019 (8) |
| C6 | 0.0192 (11) | 0.0309 (11) | 0.0233 (11) | 0.0017 (8) | 0.0016 (9) | 0.0021 (9) |
| C7 | 0.0156 (10) | 0.0176 (9) | 0.0187 (10) | 0.0007 (7) | −0.0008 (8) | −0.0007 (8) |
| C8 | 0.0201 (10) | 0.0191 (10) | 0.0189 (10) | 0.0000 (8) | −0.0009 (8) | 0.0035 (8) |
| C9 | 0.0163 (10) | 0.0163 (9) | 0.0206 (10) | 0.0006 (7) | −0.0019 (8) | −0.0007 (8) |
| C91 | 0.0245 (11) | 0.0217 (10) | 0.0284 (12) | 0.0074 (8) | 0.0011 (9) | 0.0066 (9) |
Geometric parameters (Å, º) top
| S1—O1 | 1.4322 (14) | C5—H5 | 0.9500 |
| S1—O2 | 1.4416 (14) | C6—H6 | 0.9500 |
| S1—C4 | 1.772 (2) | C7—C8 | 1.392 (3) |
| S1—C7 | 1.782 (2) | C7—C9i | 1.399 (3) |
| Cl1—C1 | 1.738 (2) | C8—C9 | 1.386 (3) |
| C1—C2 | 1.384 (3) | C8—H8 | 0.9500 |
| C1—C6 | 1.387 (3) | C9—C7i | 1.399 (3) |
| C2—C3 | 1.389 (3) | C9—C91 | 1.507 (3) |
| C2—H2 | 0.9500 | C91—H91A | 0.9800 |
| C3—C4 | 1.388 (3) | C91—H91B | 0.9800 |
| C4—C5 | 1.389 (3) | C91—H91C | 0.9800 |
| C5—C6 | 1.384 (3) | | |
| | | |
| O1—S1—O2 | 118.67 (9) | C4—C5—H5 | 120.2 |
| O1—S1—C4 | 108.12 (9) | C5—C6—C1 | 118.95 (19) |
| O2—S1—C4 | 107.72 (9) | C5—C6—H6 | 120.5 |
| O1—S1—C7 | 110.17 (9) | C1—C6—H6 | 120.5 |
| O2—S1—C7 | 107.60 (9) | C8—C7—C9i | 121.76 (18) |
| C4—S1—C7 | 103.49 (9) | C8—C7—S1 | 115.96 (15) |
| C2—C1—C6 | 122.01 (19) | C9i—C7—S1 | 122.24 (14) |
| C2—C1—Cl1 | 118.91 (16) | C9—C8—C7 | 122.29 (18) |
| C6—C1—Cl1 | 119.08 (16) | C9—C8—H8 | 118.9 |
| C1—C2—C3 | 118.82 (18) | C7—C8—H8 | 118.9 |
| C1—C2—H2 | 120.6 | C8—C9—C7i | 115.95 (17) |
| C3—C2—H2 | 120.6 | C7i—C9—C91 | 124.97 (18) |
| C4—C3—C2 | 119.53 (19) | C8—C9—C91 | 119.06 (18) |
| C4—C3—H3 | 120.2 | C9—C91—H91A | 109.5 |
| C2—C3—H3 | 120.2 | C9—C91—H91B | 109.5 |
| C3—C4—C5 | 121.12 (19) | H91A—C91—H91B | 109.5 |
| C3—C4—S1 | 119.65 (16) | C9—C91—H91C | 109.5 |
| C5—C4—S1 | 119.21 (14) | H91A—C91—H91C | 109.5 |
| C6—C5—C4 | 119.56 (18) | H91B—C91—H91C | 109.5 |
| C6—C5—H5 | 120.2 | | |
| | | |
| C6—C1—C2—C3 | 1.6 (3) | C4—C5—C6—C1 | −0.2 (3) |
| Cl1—C1—C2—C3 | −178.89 (15) | C2—C1—C6—C5 | −0.8 (3) |
| C1—C2—C3—C4 | −1.2 (3) | Cl1—C1—C6—C5 | 179.62 (15) |
| C2—C3—C4—C5 | 0.2 (3) | O1—S1—C7—C8 | 148.45 (15) |
| C2—C3—C4—S1 | −177.68 (15) | O2—S1—C7—C8 | 17.70 (17) |
| O1—S1—C4—C3 | −4.3 (2) | C4—S1—C7—C8 | −96.15 (16) |
| O2—S1—C4—C3 | 125.05 (17) | O1—S1—C7—C9i | −33.78 (19) |
| C7—S1—C4—C3 | −121.19 (17) | O2—S1—C7—C9i | −164.53 (16) |
| O1—S1—C4—C5 | 177.70 (15) | C4—S1—C7—C9i | 81.62 (17) |
| O2—S1—C4—C5 | −52.91 (18) | C9i—C7—C8—C9 | 0.1 (3) |
| C7—S1—C4—C5 | 60.85 (17) | S1—C7—C8—C9 | 177.89 (15) |
| C3—C4—C5—C6 | 0.5 (3) | C7—C8—C9—C7i | −0.1 (3) |
| S1—C4—C5—C6 | 178.43 (15) | C7—C8—C9—C91 | −178.32 (18) |
| Symmetry code: (i) −x+1, −y+1, −z+1. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C2—H2···O2ii | 0.95 | 2.54 | 3.254 (3) | 132 |
| C3—H3···O1 | 0.95 | 2.52 | 2.910 (3) | 105 |
| C8—H8···O2 | 0.95 | 2.42 | 2.859 (2) | 108 |
| Symmetry code: (ii) x, y−1, z. |
Experimental details
| Crystal data |
| Chemical formula | C20H16Cl2O4S2 |
| Mr | 455.35 |
| Crystal system, space group | Triclinic, P1 |
| Temperature (K) | 150 |
| a, b, c (Å) | 5.4859 (2), 6.9595 (4), 12.5211 (7) |
| α, β, γ (°) | 98.6245 (17), 93.561 (4), 92.174 (3) |
| V (Å3) | 471.18 (4) |
| Z | 1 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.59 |
| Crystal size (mm) | 0.10 × 0.10 × 0.03 |
| |
| Data collection |
| Diffractometer | KappaCCD
diffractometer |
| Absorption correction | Multi-scan
(SORTAV; Blessing, 1995, 1997) |
| Tmin, Tmax | 0.943, 0.985 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 5965, 2112, 1689 |
| Rint | 0.039 |
| (sin θ/λ)max (Å−1) | 0.649 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.039, 0.104, 1.07 |
| No. of reflections | 2112 |
| No. of parameters | 128 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.39, −0.53 |
Selected geometric parameters (Å, º) top| S1—O1 | 1.4322 (14) | S1—C7 | 1.782 (2) |
| S1—O2 | 1.4416 (14) | Cl1—C1 | 1.738 (2) |
| S1—C4 | 1.772 (2) | | |
| | | |
| O1—S1—O2 | 118.67 (9) | C8—C7—C9i | 121.76 (18) |
| O1—S1—C4 | 108.12 (9) | C8—C7—S1 | 115.96 (15) |
| O2—S1—C4 | 107.72 (9) | C9i—C7—S1 | 122.24 (14) |
| O1—S1—C7 | 110.17 (9) | C8—C9—C7i | 115.95 (17) |
| O2—S1—C7 | 107.60 (9) | C7i—C9—C91 | 124.97 (18) |
| C4—S1—C7 | 103.49 (9) | C8—C9—C91 | 119.06 (18) |
| | | |
| O1—S1—C4—C3 | −4.3 (2) | O2—S1—C7—C8 | 17.70 (17) |
| Symmetry code: (i) −x+1, −y+1, −z+1. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C2—H2···O2ii | 0.95 | 2.54 | 3.254 (3) | 132 |
| Symmetry code: (ii) x, y−1, z. |


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2000/07/00/gg1006/gg1006fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2000/07/00/gg1006/gg1006fig2thm.gif)




In molecular solids which contain an excess of hard hydrogen-bond acceptors over hard hydrogen-bond donors (Braga et al., 1995), it is to be expected that C—H bonds, particularly those in aromatic systems, will participate as donors in hydrogen bonds of the types C—H···O or C—H···N (Hanton et al., 1992). Aromatic sulfones are particularly effective in C—H···O hydrogen-bond formation between aromatic C—H bonds acting as hydrogen-bond donors and sulfone S=O bonds acting as acceptors (Glidewell et al., 1995; Meehan et al., 1997; Ferguson et al., 1999). In particular, the bis(sulfones) (PhSO2)2CH2 (Glidewell et al., 1995) and (4-MeOC6H4SO2)2CH2 (Meehan et al., 1997) form one-dimensional chains and a three-dimensional framework, respectively, as a result of such hydrogen-bond formation. As an example of a bis(sulfone) in which a further aromatic nucleus has been incorporated between the two sulfone groups, we now report the structure of the title compound, (I), wherein the molecules are linked into a molecular ladder by C—H···O hydrogen bonds. \sch
Compound (I) crystallizes in the space group P1 with Z = 1; hence the molecules lie across centres of inversion and, for the sake of convenience, the reference molecule has been placed across the inversion centre at (1/2, 1/2, 1/2). The molecular conformation of (I) (Fig. 1) is dominated by the near coplanarity of the O—S—C—C fragments (Table 1). As commonly observed in diaryl sulfones (Ferguson et al., 1999), there is an electrostatic attraction between the S=O bond, which is polarized in the sense S+—O−, and an ortho C—H bond in the adjacent ring, polarized in the sense C−—H+. In the most common arrangement, as also found here, each O atom of the sulfone unit interacts with a different ring, so that the local symmetry around the S atom is close to C2.
Both of the S1—C7—C9i and C7i—C9—C91 bond angles [symmetry code: (i) 1 − x, 1 − y, 1 − z] are significantly greater than 120° (Table 1). This type of angular distortion has been observed in several diaryl sulfones where there is a methyl group adjacent to the SO2 unit (Jeyakanthan et al., 1998), and it may plausibly be interpreted as arising from repulsive interactions between the methyl group and the adjacent sulfone unit. The O—S—O bond angle is much larger than tetrahedral. All the bond lengths are typical of their types (Allen et al., 1987).
There is a single type of intermolecular hydrogen bond (Table 2) linking the molecules. Atom C2 at (x, y, z), a component of the molecule centred at (1/2, 1/2, 1/2), acts as hydrogen-bond donor to O2 at (x, −1 + y, z), which is a component of the molecule centred at (1/2, −1/2, 1/2): repetition of this hydrogen bond generates a C(6) chain running parallel to [010] and generated by translation (Fig. 2). The symmetry-related C2 in the original (1/2, 1/2, 1/2) molecule is at (1 − x, 1 − y, 1 − z), and this acts as donor to O2 at (1 − x, 2 − y, 1 − z), which is a component of the molecule centred at (1/2, 3/2, 1/2); hence another C(6) chain is generated, running antiparallel to the first. The overall supramolecular structure (Fig. 2) thus takes the form of a molecular ladder, in which the uprights are formed by a pair of antiparallel C(6) chains, while the rungs are provided by the central aryl rings of successive molecules. Between the rungs are R22(22) rings centred at (1/2, n, 1/2) (n = zero or integer), but the aromatic and methyl H atoms preclude the presence of any void space. A single ladder runs through each unit cell, and there are no significant interactions between neighbouring ladders; in particular, there are no aromatic π···π stacking interactions.
It is notable that the intermolecular hydrogen bonding does not involve the central aryl ring. By contrast, in each of (PhSO2)2CH2 (Glidewell et al., 1995) and (4-MeOC6H4SO2)2CH2 (Meehan et al., 1997), both of the C—H bonds of the central methylene unit are engaged in hydrogen-bond formation. It is thus of interest to consider some related compounds retrieved from the Cambridge Structural Database (CSD; Allen & Kennard, 1993). In (PhSO2CH2)2 (FAHNOR; Hauback & Mo, 1990), although the molecules could lie across centres of inversion, as in (I), they do not in fact do so: the molecules are again linked into molecular ladders, but the C—H···O hydrogen bonds forming the uprights use only H atoms from the central –CH2—CH2– unit. The ladders are linked into a two-dimensional array by further C—H···O hydrogen bonds involving the aromatic rings. For neither the Z isomer of PhSO2CH═CHSO2Ph (DEBYIS; De Lucchi et al., 1985) nor the centrosymmetric E isomer (DEBYOY; De Lucchi et al., 1985) are there any H atom coordinates in the CSD; nonetheless, it is clear from the short intermolecular C···O distances that the intermolecular aggregation is dominated by the aromatic C—H bonds rather than those in the alkene fragment. The factors which determine the involvement of aromatic versus other C—H bonds in these systems are as yet not fully clear. However, it may be expected that aliphatic C—H bonds adjacent to SO2 units are more acidic than is typical for aliphatic C—H bonds, and thus more like aromatic C—H bonds in their hydrogen-bonding behaviour.