The crystal structure of the title compound, [FeCl
2(C
4H
8O
2)(H
2O)
2]
n, contains six-coordinate Fe
II atoms in approximately octahedral environments. The Fe
II atoms have
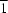
symmetry,
i.e. all pairs of identical ligands are
trans. The structure consists of polymeric chains made up of dioxane molecules, in the chair conformation with
symmetry, linking the Fe
II centers. The chains are crosslinked by O—H

Cl hydrogen bonds.
Supporting information
CCDC reference: 193402
Anhydrous iron(II) chloride (0.50 g) and lithium chloride (0.50 g) were placed
in a 100 ml beaker. 1,4-Dioxane (50 ml) was added and the solution was heated
and stirred for 2 h. After filtering off unreacted starting material, the
resulting solution was separated in 5 ml vials for evaporation and
crystallization.
Data collection: SMART (Bruker, 1999); cell refinement: SAINT (Bruker, 1999); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: SHELXL97; software used to prepare material for publication: SHELXL97.
catena-diaqua-dichloro-(µ-1,4-dioxane-O,
O')-iron(II)
top
Crystal data top
| [FeCl2(C4H8O2)(H2O)2] | F(000) = 256 |
| Mr = 250.89 | Dx = 1.811 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| a = 6.8590 (18) Å | Cell parameters from 1309 reflections |
| b = 9.273 (2) Å | θ = 3.3–29.2° |
| c = 7.925 (2) Å | µ = 2.19 mm−1 |
| β = 114.082 (5)° | T = 100 K |
| V = 460.2 (2) Å3 | Irregular, red–brown |
| Z = 2 | 0.20 × 0.20 × 0.15 mm |
Data collection top
Bruker SMART CCD
diffractometer | 1058 independent reflections |
| Radiation source: fine-focus sealed tube | 965 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.021 |
| ω scans | θmax = 29.2°, θmin = 3.3° |
Absorption correction: empirical (using intensity measurements)
multipole expansion (Blessing, 1995; Sheldrick, 1996) | h = −9→7 |
| Tmin = 0.364, Tmax = 0.659 | k = −5→12 |
| 1799 measured reflections | l = −10→9 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.035 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.094 | All H-atom parameters refined |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.0666P)2 + 0.1903P]
where P = (Fo2 + 2Fc2)/3 |
| 1058 reflections | (Δ/σ)max < 0.001 |
| 76 parameters | Δρmax = 0.92 e Å−3 |
| 0 restraints | Δρmin = −0.67 e Å−3 |
Crystal data top
| [FeCl2(C4H8O2)(H2O)2] | V = 460.2 (2) Å3 |
| Mr = 250.89 | Z = 2 |
| Monoclinic, P21/n | Mo Kα radiation |
| a = 6.8590 (18) Å | µ = 2.19 mm−1 |
| b = 9.273 (2) Å | T = 100 K |
| c = 7.925 (2) Å | 0.20 × 0.20 × 0.15 mm |
| β = 114.082 (5)° | |
Data collection top
Bruker SMART CCD
diffractometer | 1058 independent reflections |
Absorption correction: empirical (using intensity measurements)
multipole expansion (Blessing, 1995; Sheldrick, 1996) | 965 reflections with I > 2σ(I) |
| Tmin = 0.364, Tmax = 0.659 | Rint = 0.021 |
| 1799 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.035 | 0 restraints |
| wR(F2) = 0.094 | All H-atom parameters refined |
| S = 1.05 | Δρmax = 0.92 e Å−3 |
| 1058 reflections | Δρmin = −0.67 e Å−3 |
| 76 parameters | |
Special details top
Experimental. The decay correction was applied simultaneously with the absorption correction
in SADABS. No formal measure of the extent of decay is printed out by
this program. The final unit cell is obtained from the refinement of the XYZ
weighted centroids of reflections above 15 σ(I). |
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Fe1 | 0.0000 | 0.5000 | 0.0000 | 0.01226 (17) | |
| Cl1 | 0.03854 (8) | 0.23836 (5) | −0.02603 (7) | 0.01541 (18) | |
| O2 | 0.1774 (3) | 0.48894 (17) | 0.2862 (3) | 0.0173 (4) | |
| H1A | 0.240 (6) | 0.561 (4) | 0.343 (5) | 0.031 (9)* | |
| H1B | 0.268 (7) | 0.425 (4) | 0.327 (5) | 0.033 (9)* | |
| O1 | 0.2847 (3) | 0.54151 (18) | −0.0509 (2) | 0.0168 (4) | |
| C1 | 0.3476 (4) | 0.4532 (2) | −0.1708 (3) | 0.0172 (4) | |
| H2A | 0.225 (5) | 0.388 (3) | −0.237 (4) | 0.016 (7)* | |
| H2B | 0.379 (6) | 0.518 (3) | −0.254 (5) | 0.026 (9)* | |
| C2 | 0.4591 (4) | 0.6350 (2) | 0.0583 (4) | 0.0178 (5) | |
| H3A | 0.492 (5) | 0.697 (4) | −0.028 (4) | 0.022 (8)* | |
| H3B | 0.406 (5) | 0.690 (3) | 0.140 (4) | 0.022 (7)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Fe1 | 0.0110 (3) | 0.0113 (3) | 0.0139 (3) | 0.00039 (15) | 0.00445 (19) | 0.00011 (14) |
| Cl1 | 0.0147 (3) | 0.0115 (3) | 0.0183 (3) | 0.00052 (17) | 0.0050 (2) | −0.00036 (17) |
| O2 | 0.0167 (9) | 0.0136 (7) | 0.0181 (9) | 0.0005 (6) | 0.0034 (7) | −0.0013 (6) |
| O1 | 0.0125 (8) | 0.0181 (7) | 0.0211 (8) | −0.0019 (6) | 0.0082 (6) | −0.0052 (6) |
| C1 | 0.0164 (11) | 0.0201 (10) | 0.0167 (10) | −0.0007 (9) | 0.0084 (9) | −0.0028 (9) |
| C2 | 0.0149 (11) | 0.0166 (11) | 0.0230 (11) | −0.0030 (8) | 0.0090 (9) | −0.0026 (8) |
Geometric parameters (Å, º) top
| Fe1—O1 | 2.1830 (17) | O1—C2 | 1.444 (3) |
| Fe1—O1i | 2.1830 (17) | O1—C1 | 1.448 (3) |
| Fe1—O2i | 2.0934 (19) | C1—C2ii | 1.502 (3) |
| Fe1—O2 | 2.0934 (19) | C1—H2A | 0.99 (3) |
| Fe1—Cl1i | 2.4583 (8) | C1—H2B | 0.98 (3) |
| Fe1—Cl1 | 2.4583 (8) | C2—C1ii | 1.502 (3) |
| O2—H1A | 0.82 (4) | C2—H3A | 0.99 (3) |
| O2—H1B | 0.83 (4) | C2—H3B | 1.00 (3) |
| | | |
| O1—Fe1—O1i | 180.0 | H1A—O2—H1B | 103 (4) |
| O1—Fe1—Cl1i | 88.65 (5) | C2—O1—C1 | 109.67 (18) |
| O1i—Fe1—Cl1i | 91.35 (5) | C2—O1—Fe1 | 125.44 (13) |
| O1—Fe1—Cl1 | 91.35 (5) | C1—O1—Fe1 | 123.40 (14) |
| O1i—Fe1—Cl1 | 88.65 (5) | O1—C1—C2ii | 110.2 (2) |
| O2i—Fe1—O2 | 180.0 | O1—C1—H2A | 105.9 (17) |
| O2i—Fe1—O1 | 87.69 (7) | C2ii—C1—H2A | 109.5 (16) |
| O2—Fe1—O1 | 92.31 (7) | O1—C1—H2B | 107.4 (19) |
| O2i—Fe1—O1i | 92.31 (7) | C2ii—C1—H2B | 110 (2) |
| O2—Fe1—O1i | 87.69 (7) | H2A—C1—H2B | 113 (3) |
| O2i—Fe1—Cl1i | 90.44 (4) | O1—C2—C1ii | 110.13 (18) |
| O2—Fe1—Cl1i | 89.56 (4) | O1—C2—H3A | 107.6 (18) |
| O2i—Fe1—Cl1 | 89.56 (4) | C1ii—C2—H3A | 109.5 (18) |
| O2—Fe1—Cl1 | 90.44 (4) | O1—C2—H3B | 105.1 (19) |
| Cl1i—Fe1—Cl1 | 180.0 | C1ii—C2—H3B | 111.1 (18) |
| Fe1—O2—H1A | 120 (3) | H3A—C2—H3B | 113 (3) |
| Fe1—O2—H1B | 118 (3) | | |
| Symmetry codes: (i) −x, −y+1, −z; (ii) −x+1, −y+1, −z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H1A···Cl1iii | 0.82 (4) | 2.31 (4) | 3.1202 (19) | 171 (4) |
| O2—H1B···Cl1iv | 0.83 (4) | 2.31 (4) | 3.1280 (19) | 173 (3) |
| Symmetry codes: (iii) −x+1/2, y+1/2, −z+1/2; (iv) x+1/2, −y+1/2, z+1/2. |
Experimental details
| Crystal data |
| Chemical formula | [FeCl2(C4H8O2)(H2O)2] |
| Mr | 250.89 |
| Crystal system, space group | Monoclinic, P21/n |
| Temperature (K) | 100 |
| a, b, c (Å) | 6.8590 (18), 9.273 (2), 7.925 (2) |
| β (°) | 114.082 (5) |
| V (Å3) | 460.2 (2) |
| Z | 2 |
| Radiation type | Mo Kα |
| µ (mm−1) | 2.19 |
| Crystal size (mm) | 0.20 × 0.20 × 0.15 |
| |
| Data collection |
| Diffractometer | Bruker SMART CCD
diffractometer |
| Absorption correction | Empirical (using intensity measurements)
multipole expansion (Blessing, 1995; Sheldrick, 1996) |
| Tmin, Tmax | 0.364, 0.659 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 1799, 1058, 965 |
| Rint | 0.021 |
| (sin θ/λ)max (Å−1) | 0.686 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.035, 0.094, 1.05 |
| No. of reflections | 1058 |
| No. of parameters | 76 |
| H-atom treatment | All H-atom parameters refined |
| Δρmax, Δρmin (e Å−3) | 0.92, −0.67 |
Selected geometric parameters (Å, º) top| Fe1—O1 | 2.1830 (17) | Fe1—Cl1 | 2.4583 (8) |
| Fe1—O2 | 2.0934 (19) | | |
| | | |
| O1—Fe1—O1i | 180.0 | O2—Fe1—O1 | 92.31 (7) |
| O1—Fe1—Cl1i | 88.65 (5) | O2—Fe1—Cl1i | 89.56 (4) |
| O1—Fe1—Cl1 | 91.35 (5) | O2—Fe1—Cl1 | 90.44 (4) |
| O2i—Fe1—O2 | 180.0 | Cl1i—Fe1—Cl1 | 180.0 |
| O2i—Fe1—O1 | 87.69 (7) | | |
| Symmetry code: (i) −x, −y+1, −z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H1A···Cl1ii | 0.82 (4) | 2.31 (4) | 3.1202 (19) | 171 (4) |
| O2—H1B···Cl1iii | 0.83 (4) | 2.31 (4) | 3.1280 (19) | 173 (3) |
| Symmetry codes: (ii) −x+1/2, y+1/2, −z+1/2; (iii) x+1/2, −y+1/2, z+1/2. |


 journal menu
journal menu metal-organic compounds
metal-organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2002/08/00/fr1377/fr1377fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2002/08/00/fr1377/fr1377fig2thm.gif)




For many years, crystal engineering has utilized the hydrogen bond for the formation of extended networks, thus creating structures with a variety of pore shapes and sizes able to accommodate various guest molecules. Many of the networks that have been reported are neutral. An obvious alternative to neutral frameworks are ionic ones, in particular, cationic frameworks. These could then play host to a variety of negatively charged species of various sizes. It has been suggested that a metal-cation–dioxane network could achieve this (Hasch et al., 2000). A number of structures have been reported involving alkali-metal–dioxane networks (Taube et al., 1993; Eaborn et al., 1997; Kühl et al., 1999, 2000; Hasche et al., 2000). These structures indicate the variety of structures obtainable as a result of variations in the coordination at the metal center; thus, one-dimensional chains, two-dimensional sheets, and three-dimensional networks have all been observed. The only other known extended dioxane–metal structures are chains with hard metal centers, for example, AlIII (Boardman et al., 1983), GaIII (Boardman et al., 1984), MgII (Parvez et al., 1988), CdII (Almond et al., 1991), TlIII (Jeffs et al., 1983), NdIII (Taube et al., 1996), and FeII (Müller et al., 1997). Recently, another type of chain structure has been reported where 1,4-dioxane links 1,2-diiodotetrafluoroethane molecules by coordination to the two I atoms (Chu et al., 2001).
While attempting to extend our work on lithium-containing cationic networks to include iron(II)-containing anions by the reaction of LiCl and FeCl2 in dioxane, only the lithium-free title compound, (I), a one-dimensional polymeric structure, was obtained. This compound contains a pseudo-octahedral six-coordinate FeII cation with 1 symmetry, coordinated by two trans Cl- ions and two trans water molecules. The final two coordination sites are occupied by 1,4-dioxane rings bound through O atoms, as shown in Fig. 1. The O atoms of the dioxane ring, which adopts a chair conformation, bind to two FeII atoms on either side, with a bond distance of 2.1830 (17) Å. This coordination results in the formation of chains parallel to a and these chains build the backbone of the crystal structure, similar to the case in catena-[[bis(2,2,6,6-tetramethylheptane-3,5-dionato)iron(II)]-µ-1,4-dioxane] (Müller et al., 1997). In the present case, the iron–dioxane chains are crosslinked by Owater···Cl hydrogen bonding (Fig. 2). The hydrogen bonding involves the H atoms located on the coordinated water molecules and the coordinated Cl- ions on the FeII atoms of two adjacent chains. Each water molecule hydrogen bonds to two Cl- ions from two adjacent FeII centers, and each chloride ion accepts two hydrogen bonds from two adjacent FeII centers, i.e. an approximately square arrangement with the two Cl and two O atoms at opposite corners, each side representing a hydrogen bond, and each corner bound to a different FeII atom; an approximately planar FeOClH layer is thus formed perpendicular to the Fe—Odioxane vector within the polymer chain.