The title complex, {[Ni(C
2H
8N
2)
3][Na(NCS)
3(H
2O)]}
n, consists of discrete [Ni(en)
3]
2+ dications (en is ethylenediamine) and polymeric [(H
2O)
0.5Na(NCS)
3(H
2O)
0.5]
n2n− anions. The compound crystallizes in space group
P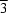 c
c1. The Ni
II atom lies on a threefold axis and has a distorted octahedral coordination geometry. The Na
+ cation also lies on a site with imposed crystallographic threefold symmetry and is coordinated by the thiocyanate N atoms (the thiocyanates are in general positions), by one water molecule with crystallographically imposed 32 symmetry and by a second water molecule with crystallographically imposed
symmetry. The unique Na atom thus has trigonal–bipyramidal coordination. The O atoms of the water molecules bridge the Na
+ cations to form one-dimensional polymeric chains in the crystal structure. The [Ni(en)
3]
2+ dications are distributed around and between the chains and are linked to them
via N—H

S hydrogen bonds.
Supporting information
CCDC reference: 167817
To an aqueous solution (15 ml) of Ni(NO3)2·6H2O (0.37 g, 1 mmol) and
ethylenediamine (0.18 g, 3 mmol), an aqueous solution (15 ml) containing NaSCN
(0.24 g, 3 mmol) was added with stirring. After stirring for 30 min at room
temperature, the purple solution was filtered. Purple single crystals of (I)
were obtained from the solution by slow evaporation of the solvent over a
week.
The systematic absences and Laue symmetry allowed the space group to be either
P3c1 or P3c1; P3c1 was selected and
confirmed by the analysis. The eight H atoms of the unique en ligand were
allowed for as riding atoms (C—H = 0.97 Å and N—H = 0.90 Å). It was
not possible to locate the H atoms of the water molecules on the 32 and 3
special positions.
Data collection: XSCANS (Siemens, 1991); cell refinement: XSCANS; data reduction: SHELXTL-Plus (Sheldrick, 1990a); program(s) used to solve structure: SHELXS97 (Sheldrick, 1990b); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: SHELXTL-Plus; software used to prepare material for publication: SHELXTL-Plus.
catena-Poly[tris(ethylenediamine-
κ2N)nickel(II)
[[trithiocyanato-
κ3N-sodium(I)]-µ-aqua-
κ2O:
O]]
top
Crystal data top
| [Ni(C2H8N2)3][Na(NCS)3(H2O)] | Dx = 1.511 Mg m−3 |
| Mr = 454.27 | Mo Kα radiation, λ = 0.71073 Å |
| Trigonal, P3c1 | Cell parameters from 22 reflections |
| Hall symbol: -P 3 2"c | θ = 3.1–16.1° |
| a = 11.588 (1) Å | µ = 1.32 mm−1 |
| c = 17.177 (2) Å | T = 293 K |
| V = 1997.5 (3) Å3 | Prism, purple |
| Z = 4 | 0.48 × 0.46 × 0.40 mm |
| F(000) = 952 | |
Data collection top
Siemens P4
diffractometer | 1001 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.025 |
| Graphite monochromator | θmax = 27.0°, θmin = 2.0° |
| ω scans | h = 0→14 |
Absorption correction: empirical (using intensity measurements)
(North et al., 1968) | k = −14→1 |
| Tmin = 0.552, Tmax = 0.589 | l = 0→21 |
| 3084 measured reflections | 3 standard reflections every 97 reflections |
| 1462 independent reflections | intensity decay: 1.8% |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.038 | H-atom parameters constrained |
| wR(F2) = 0.130 | w = 1/[σ2(Fo2) + (0.068P)2 + 1.1204P]
where P = (Fo2 + 2Fc2)/3 |
| S = 1.02 | (Δ/σ)max < 0.001 |
| 1462 reflections | Δρmax = 0.50 e Å−3 |
| 75 parameters | Δρmin = −0.29 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97 (Sheldrick, 1997), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0111 (14) |
Crystal data top
| [Ni(C2H8N2)3][Na(NCS)3(H2O)] | Z = 4 |
| Mr = 454.27 | Mo Kα radiation |
| Trigonal, P3c1 | µ = 1.32 mm−1 |
| a = 11.588 (1) Å | T = 293 K |
| c = 17.177 (2) Å | 0.48 × 0.46 × 0.40 mm |
| V = 1997.5 (3) Å3 | |
Data collection top
Siemens P4
diffractometer | 1001 reflections with I > 2σ(I) |
Absorption correction: empirical (using intensity measurements)
(North et al., 1968) | Rint = 0.025 |
| Tmin = 0.552, Tmax = 0.589 | 3 standard reflections every 97 reflections |
| 3084 measured reflections | intensity decay: 1.8% |
| 1462 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.038 | 0 restraints |
| wR(F2) = 0.130 | H-atom parameters constrained |
| S = 1.02 | Δρmax = 0.50 e Å−3 |
| 1462 reflections | Δρmin = −0.29 e Å−3 |
| 75 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Ni | 0.6667 | 0.3333 | 0.37758 (3) | 0.0361 (3) | |
| Na | 1.0000 | 0.0000 | 0.37356 (10) | 0.0324 (4) | |
| S | 0.52065 (11) | −0.13980 (11) | 0.36888 (5) | 0.0669 (4) | |
| O1 | 1.0000 | 0.0000 | 0.2500 | 0.086 (2) | |
| O2 | 1.0000 | 0.0000 | 0.5000 | 0.0611 (16) | |
| N1 | 0.8306 (3) | 0.3599 (3) | 0.30894 (14) | 0.0475 (7) | |
| H1A | 0.8067 | 0.3456 | 0.2585 | 0.057* | |
| H1B | 0.8984 | 0.4441 | 0.3139 | 0.057* | |
| N2 | 0.7250 (3) | 0.2179 (3) | 0.44568 (14) | 0.0489 (7) | |
| H2A | 0.7226 | 0.2348 | 0.4966 | 0.059* | |
| H2B | 0.6693 | 0.1305 | 0.4373 | 0.059* | |
| N3 | 0.7892 (5) | −0.0666 (5) | 0.3746 (3) | 0.1108 (16) | |
| C1 | 0.8732 (4) | 0.2664 (4) | 0.33454 (19) | 0.0604 (10) | |
| H1C | 0.9646 | 0.2983 | 0.3187 | 0.072* | |
| H1D | 0.8172 | 0.1798 | 0.3110 | 0.072* | |
| C2 | 0.8622 (4) | 0.2550 (4) | 0.4223 (2) | 0.0602 (10) | |
| H2C | 0.8826 | 0.1878 | 0.4401 | 0.072* | |
| H2D | 0.9254 | 0.3394 | 0.4459 | 0.072* | |
| C3 | 0.6769 (5) | −0.0970 (4) | 0.3715 (2) | 0.0666 (11) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Ni | 0.0398 (3) | 0.0398 (3) | 0.0285 (3) | 0.01992 (15) | 0.000 | 0.000 |
| Na | 0.0272 (6) | 0.0272 (6) | 0.0427 (10) | 0.0136 (3) | 0.000 | 0.000 |
| S | 0.0730 (7) | 0.0755 (7) | 0.0499 (5) | 0.0355 (6) | −0.0101 (4) | 0.0023 (5) |
| O1 | 0.102 (4) | 0.102 (4) | 0.053 (4) | 0.0509 (19) | 0.000 | 0.000 |
| O2 | 0.067 (2) | 0.067 (2) | 0.050 (3) | 0.0333 (12) | 0.000 | 0.000 |
| N1 | 0.0514 (17) | 0.0565 (18) | 0.0376 (12) | 0.0292 (14) | 0.0049 (12) | 0.0044 (12) |
| N2 | 0.0569 (17) | 0.0529 (16) | 0.0374 (12) | 0.0279 (15) | −0.0007 (12) | 0.0035 (12) |
| N3 | 0.074 (3) | 0.081 (3) | 0.179 (5) | 0.039 (2) | 0.007 (3) | 0.005 (3) |
| C1 | 0.074 (3) | 0.081 (3) | 0.0471 (18) | 0.054 (2) | 0.0100 (18) | 0.0084 (18) |
| C2 | 0.067 (2) | 0.081 (3) | 0.0505 (19) | 0.050 (2) | −0.0030 (17) | 0.0057 (18) |
| C3 | 0.075 (3) | 0.044 (2) | 0.081 (3) | 0.030 (2) | 0.005 (2) | −0.0004 (18) |
Geometric parameters (Å, º) top
| Ni—N1 | 2.123 (3) | C1—C2 | 1.514 (5) |
| Ni—N2 | 2.125 (3) | N1—H1A | 0.90 |
| Na—O1 | 2.1224 (18) | N1—H1B | 0.90 |
| Na—O2 | 2.1719 (18) | N2—H2A | 0.90 |
| Na—N3 | 2.163 (5) | N2—H2B | 0.90 |
| S—C3 | 1.621 (5) | C1—H1C | 0.97 |
| N1—C1 | 1.465 (4) | C1—H1D | 0.97 |
| N2—C2 | 1.480 (5) | C2—H2C | 0.97 |
| N3—C3 | 1.167 (5) | C2—H2D | 0.97 |
| | | |
| N1—Ni—N2 | 81.61 (10) | Ni—N1—H1B | 109.7 |
| N1—Ni—N1i | 92.15 (10) | Ni—N1—H1A | 109.7 |
| N1ii—Ni—N2 | 94.28 (11) | H1A—N1—H1B | 108.2 |
| N1—Ni—N2ii | 171.19 (10) | C1—N1—H1B | 109.8 |
| N1—Ni—N2i | 94.28 (11) | Ni—N2—H2B | 110.2 |
| N2—Ni—N2i | 92.62 (10) | H2A—N2—H2B | 108.5 |
| O1—Na—N3 | 90.46 (16) | Ni—N2—H2A | 110.1 |
| O2—Na—N3 | 89.54 (16) | C2—N2—H2B | 110.2 |
| N3—Na—N3iii | 120.0 (2) | C2—N2—H2A | 110.3 |
| O1—Na—O2 | 180.0 | N1—C1—H1C | 110.0 |
| Na—O1—Naiv | 180.0 | N1—C1—H1D | 110.0 |
| Nav—O2—Na | 180.0 | C2—C1—H1C | 109.9 |
| C1—N1—Ni | 109.5 (2) | C2—C1—H1D | 110.0 |
| C2—N2—Ni | 107.5 (2) | H1C—C1—H1D | 108.4 |
| C3—N3—Na | 175.8 (5) | N2—C2—H2C | 109.8 |
| N1—C1—C2 | 108.6 (3) | N2—C2—H2D | 109.8 |
| N2—C2—C1 | 109.1 (3) | C1—C2—H2C | 109.9 |
| N3—C3—S | 178.9 (5) | C1—C2—H2D | 109.9 |
| C1—N1—H1A | 109.9 | H2C—C2—H2D | 108.3 |
| Symmetry codes: (i) −y+1, x−y, z; (ii) −x+y+1, −x+1, z; (iii) −y+1, x−y−1, z; (iv) x−y, −y, −z+1/2; (v) −x+2, −y, −z+1. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···Svi | 0.90 | 2.83 | 3.504 (3) | 132 |
| N1—H1B···Si | 0.90 | 2.83 | 3.681 (3) | 159 |
| N2—H2A···Svii | 0.90 | 2.72 | 3.518 (3) | 148 |
| N2—H2B···S | 0.90 | 2.96 | 3.836 (3) | 165 |
| Symmetry codes: (i) −y+1, x−y, z; (vi) y+1, x, −z+1/2; (vii) y+1, −x+y+1, −z+1. |
Experimental details
| Crystal data |
| Chemical formula | [Ni(C2H8N2)3][Na(NCS)3(H2O)] |
| Mr | 454.27 |
| Crystal system, space group | Trigonal, P3c1 |
| Temperature (K) | 293 |
| a, c (Å) | 11.588 (1), 17.177 (2) |
| V (Å3) | 1997.5 (3) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 1.32 |
| Crystal size (mm) | 0.48 × 0.46 × 0.40 |
| |
| Data collection |
| Diffractometer | Siemens P4
diffractometer |
| Absorption correction | Empirical (using intensity measurements)
(North et al., 1968) |
| Tmin, Tmax | 0.552, 0.589 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 3084, 1462, 1001 |
| Rint | 0.025 |
| (sin θ/λ)max (Å−1) | 0.639 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.038, 0.130, 1.02 |
| No. of reflections | 1462 |
| No. of parameters | 75 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.50, −0.29 |
Selected geometric parameters (Å, º) top| Ni—N1 | 2.123 (3) | Na—O2 | 2.1719 (18) |
| Ni—N2 | 2.125 (3) | Na—N3 | 2.163 (5) |
| Na—O1 | 2.1224 (18) | | |
| | | |
| N1—Ni—N2 | 81.61 (10) | N1—Ni—N2ii | 171.19 (10) |
| N1—Ni—N1i | 92.15 (10) | O1—Na—N3 | 90.46 (16) |
| N1ii—Ni—N2 | 94.28 (11) | | |
| Symmetry codes: (i) −y+1, x−y, z; (ii) −x+y+1, −x+1, z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···Siii | 0.90 | 2.83 | 3.504 (3) | 132 |
| N1—H1B···Si | 0.90 | 2.83 | 3.681 (3) | 159 |
| N2—H2A···Siv | 0.90 | 2.72 | 3.518 (3) | 148 |
| N2—H2B···S | 0.90 | 2.96 | 3.836 (3) | 165 |
| Symmetry codes: (i) −y+1, x−y, z; (iii) y+1, x, −z+1/2; (iv) y+1, −x+y+1, −z+1. |


 journal menu
journal menu metal-organic compounds
metal-organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2002/12/00/fg1666/fg1666fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2002/12/00/fg1666/fg1666fig2thm.gif)




The Na+ cation plays an important role in the growth of organisms and exists extensively in various kinds of organisms in the form of complexes of biological function. Research into the coordination environment of the Na+ cation is conducive to further understanding of its biological function. The Na+ cation usually forms four- or six-coordinated complexes (Goher & Mautner, 1994; Paixão et al., 2000). Five-, seven- or eight-coordinated sodium complexes are relatively rare (Hauptmann et al., 1999; Farrugia & Watson, 1999; Krishnakumar et al., 2001). We report here the preparation and crystal structure of the title five-coordinated Na complex, (I), which has trigonal-bipyramidal geometry. \sch
The molecular structure of (I) is shown in Fig. 1. The complex comprises discrete [Ni(en)3]2+ cations (en is ethylenediamine) and polymeric anionic [(H2O)0.5Na(NCS)3(H2O)0.5]n2n- chains in space group P3c1. The NiII atom lies on a threefold axis and has slightly distorted octahedral geometry, being coordinated by six N atoms from three ethylenediamine ligands. All bond lengths and angles involving the Ni atom are similar to those found in comparable complexes (e.g. Huang & Huang, 1984).
The Na atom in the anion lies on a threefold axis and has trigonal-bipyramidal coordination geometry, with the equatorial positions occupied by the three N atoms from three symmetry-related thiocyanate anions and the axial positions by the O atoms of the bridging water molecules. The thiocyanate ion is in a general position. Water atom O1 lies on a site with crystallographic 32 symmetry, while water atom O2 lies on a site with 3 crystallographic symmetry.
The N—Na—N angles are 120 (2)° and the Na—O2 bond [2.1719 (18) Å] is slightly longer than Na—O1 [2.1224 (18) Å]. These Na—O distances in (I) are significantly shorter than the Na—O(bridging aqua) distances in other water O-bridged complexes, e.g. [NaMn(pyz)(N3)(H2O)] [2.455 (2) Å; pyz is pyrazine; Goher et al., 1993] and [NaCu{C6H3(COO)3}(H2O)4]·2H2O [2.469 (2) Å; Chui et al., 1999]. Na—O distances of 2.366 (4) and 2.375 (4) Å were found in [NaCu(pic)2(N3)(H2O)2]n (pic is picolinate; (Goher et al.,. 1994). The Na—N—C angle in (I) is 175.8 (5)°, deviating slightly from the expected 180°. Other thiocyanate dimensions are normal.
It was not possible to locate the water H atoms, which have to be disordered because of the imposed 32 and 3 symmetry at the water O atoms. In the crystal structure of (I), the O atoms of the water molecules bridge Na+ cations to form infinite –O–Na–O–Na– chains, in which the monomer is the [(H2O)0.5Na(NCS)3(H2O)0.5]2- anion. The space-group symmetry ensures that adjacent [Na(NCS)3]2- moieties in the polymeric chains are arranged in a staggered stacking fashion along the c axis (Fig. 2). The [Ni(en)3]2+ cations connect the [(H2O)0.5Na(NCS)3(H2O)0.5]n2n- anion chains by both electrostatic and hydrogen-bond interactions, involving the N—H moieties of the en ligands and the S atoms of the thiocyanate groups, to yield a three-dimensional network.