Buy article online - an online subscription or single-article purchase is required to access this article.
Molecules of phthalimide [1
H-isoindole-1,3(2
H)-dione], C
8H
5NO
2, are linked by N-H

O hydrogen bonds [H
O 2.02 Å, N
O 2.8781 (16) Å and N-H
O 167°] and by C-H
O hydrogen bonds [H
O 2.54 and 2.56 Å, C
O 3.3874 (18) and 3.4628 (19) Å, and C-H
O 149 and 159°] into molecular ribbons, which are pierced by three different ring motifs; there are two centrosymmetric
R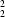
(8) rings, each containing a single hydrogen bond, N-H
O in one case and C-H
O in the other, and
R(9) rings containing all three hydrogen bonds.
Supporting information
CCDC reference: 180152
Crystals of (I) were obtained following at attempt to crystallize
N,N'-dithiodiphthalimide from hot pyridine.
Compound (I) crystallized in the monoclinic system; space group
P21/n was uniquely assigned from the systematic absences. H
atoms were treated as riding atoms with distances C—H 0.95 Å and N—H
0.88 Å.
Data collection: KappaCCD Server Software (Nonius, 1997); cell refinement: DENZO-SMN (Otwinowski & Minor, 1997); data reduction: DENZO-SMN; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: PLATON (Spek, 2001); software used to prepare material for publication: SHELXL97 (Sheldrick, 1997) and PRPKAPPA (Ferguson, 1999).
'Phthalimide [1
H-Isoindole-1,3(2
H)-dione]'
top
Crystal data top
| C8H5NO2 | F(000) = 304 |
| Mr = 147.13 | Dx = 1.510 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 1456 reflections |
| a = 3.7303 (2) Å | θ = 3.2–27.5° |
| b = 7.6638 (4) Å | µ = 0.11 mm−1 |
| c = 22.6435 (13) Å | T = 150 K |
| β = 90.369 (2)° | Prism, colourless |
| V = 647.33 (6) Å3 | 0.15 × 0.05 × 0.05 mm |
| Z = 4 | |
Data collection top
KappaCCD
diffractometer | 1456 independent reflections |
| Radiation source: fine-focus sealed X-ray tube | 1061 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.054 |
| ϕ scans, and ω scans with κ offsets | θmax = 27.5°, θmin = 3.2° |
Absorption correction: multi-scan
(DENZO-SMN; Otwinowski & Minor, 1997) | h = −4→4 |
| Tmin = 0.984, Tmax = 0.995 | k = −9→8 |
| 4750 measured reflections | l = −29→25 |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.043 | H-atom parameters constrained |
| wR(F2) = 0.103 | w = 1/[σ2(Fo2) + (0.0485P)2 + 0.0173P]
where P = (Fo2 + 2Fc2)/3 |
| S = 1.02 | (Δ/σ)max < 0.001 |
| 1456 reflections | Δρmax = 0.22 e Å−3 |
| 101 parameters | Δρmin = −0.22 e Å−3 |
| 0 restraints | Extinction correction: SHELXL, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.037 (7) |
Crystal data top
| C8H5NO2 | V = 647.33 (6) Å3 |
| Mr = 147.13 | Z = 4 |
| Monoclinic, P21/n | Mo Kα radiation |
| a = 3.7303 (2) Å | µ = 0.11 mm−1 |
| b = 7.6638 (4) Å | T = 150 K |
| c = 22.6435 (13) Å | 0.15 × 0.05 × 0.05 mm |
| β = 90.369 (2)° | |
Data collection top
KappaCCD
diffractometer | 1456 independent reflections |
Absorption correction: multi-scan
(DENZO-SMN; Otwinowski & Minor, 1997) | 1061 reflections with I > 2σ(I) |
| Tmin = 0.984, Tmax = 0.995 | Rint = 0.054 |
| 4750 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.043 | 0 restraints |
| wR(F2) = 0.103 | H-atom parameters constrained |
| S = 1.02 | Δρmax = 0.22 e Å−3 |
| 1456 reflections | Δρmin = −0.22 e Å−3 |
| 101 parameters | |
Special details top
Experimental. The program DENZO-SMN (Otwinowski & Minor, 1997) uses a scaling algorithm
[Fox, G·C. & Holmes, K·C. (1966). Acta Cryst. 20, 886–891] which
effectively corrects for absorption effects. High redundancy data were used in
the scaling program hence the 'multi-scan' code word was used. No transmission
coefficients are available from the program (only scale factors for each
frame). The scale factors in the experimental table are calculated from the
'size' command in the SHELXL97 input file. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| O1 | 0.7712 (3) | 0.99398 (13) | 0.16961 (5) | 0.0260 (3) | |
| O2 | 0.2821 (3) | 0.78244 (13) | −0.00219 (4) | 0.0257 (3) | |
| N3 | 0.5404 (3) | 0.92707 (16) | 0.07707 (5) | 0.0206 (3) | |
| C4 | 0.6202 (4) | 0.89329 (19) | 0.13625 (7) | 0.0196 (4) | |
| C5 | 0.3781 (4) | 0.78674 (19) | 0.04950 (6) | 0.0191 (4) | |
| C6 | 0.3481 (4) | 0.64822 (19) | 0.09516 (6) | 0.0177 (4) | |
| C7 | 0.2138 (4) | 0.48010 (19) | 0.09109 (7) | 0.0206 (4) | |
| C8 | 0.2283 (4) | 0.3784 (2) | 0.14201 (7) | 0.0227 (4) | |
| C9 | 0.3719 (4) | 0.4427 (2) | 0.19429 (7) | 0.0231 (4) | |
| C10 | 0.5070 (4) | 0.61196 (18) | 0.19815 (6) | 0.0198 (4) | |
| C11 | 0.4909 (4) | 0.71261 (18) | 0.14760 (6) | 0.0175 (4) | |
| H3 | 0.5882 | 1.0265 | 0.0593 | 0.025* | |
| H7 | 0.1164 | 0.4362 | 0.0552 | 0.025* | |
| H8 | 0.1378 | 0.2626 | 0.1409 | 0.027* | |
| H9 | 0.3783 | 0.3698 | 0.2282 | 0.028* | |
| H10 | 0.6058 | 0.6561 | 0.2339 | 0.024* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| O1 | 0.0328 (6) | 0.0221 (6) | 0.0230 (6) | −0.0061 (5) | −0.0054 (5) | −0.0022 (5) |
| O2 | 0.0337 (6) | 0.0251 (6) | 0.0181 (6) | −0.0055 (5) | −0.0040 (5) | −0.0002 (5) |
| N3 | 0.0280 (7) | 0.0155 (7) | 0.0182 (7) | −0.0064 (5) | −0.0021 (5) | 0.0012 (5) |
| C4 | 0.0188 (7) | 0.0206 (8) | 0.0195 (8) | −0.0002 (6) | 0.0008 (6) | −0.0010 (7) |
| C5 | 0.0185 (7) | 0.0207 (8) | 0.0180 (9) | −0.0005 (6) | 0.0008 (6) | −0.0017 (6) |
| C6 | 0.0161 (7) | 0.0185 (8) | 0.0186 (8) | 0.0008 (6) | 0.0006 (6) | 0.0001 (6) |
| C7 | 0.0185 (7) | 0.0199 (8) | 0.0234 (9) | −0.0012 (6) | −0.0002 (6) | −0.0033 (6) |
| C8 | 0.0206 (8) | 0.0164 (8) | 0.0311 (9) | 0.0002 (6) | 0.0024 (6) | 0.0007 (7) |
| C9 | 0.0212 (8) | 0.0209 (8) | 0.0273 (9) | 0.0015 (6) | 0.0029 (7) | 0.0068 (7) |
| C10 | 0.0183 (7) | 0.0212 (8) | 0.0200 (8) | 0.0018 (6) | 0.0000 (6) | −0.0002 (6) |
| C11 | 0.0154 (7) | 0.0161 (8) | 0.0212 (8) | 0.0009 (5) | 0.0015 (6) | −0.0012 (6) |
Geometric parameters (Å, º) top
| O1—C4 | 1.2158 (17) | C7—C8 | 1.393 (2) |
| O2—C5 | 1.2224 (17) | C7—H7 | 0.9500 |
| N3—C5 | 1.3813 (18) | C8—C9 | 1.387 (2) |
| N3—C4 | 1.3951 (19) | C8—H8 | 0.9500 |
| N3—H3 | 0.8800 | C9—C10 | 1.395 (2) |
| C4—C11 | 1.489 (2) | C9—H9 | 0.9500 |
| C5—C6 | 1.487 (2) | C10—C11 | 1.381 (2) |
| C6—C7 | 1.385 (2) | C10—H10 | 0.9500 |
| C6—C11 | 1.389 (2) | | |
| | | |
| C5—N3—C4 | 112.32 (12) | C8—C7—H7 | 121.5 |
| C5—N3—H3 | 123.8 | C9—C8—C7 | 121.39 (14) |
| C4—N3—H3 | 123.8 | C9—C8—H8 | 119.3 |
| O1—C4—N3 | 125.04 (14) | C7—C8—H8 | 119.3 |
| O1—C4—C11 | 129.22 (14) | C8—C9—C10 | 121.43 (14) |
| N3—C4—C11 | 105.73 (12) | C8—C9—H9 | 119.3 |
| O2—C5—N3 | 125.36 (13) | C10—C9—H9 | 119.3 |
| O2—C5—C6 | 128.60 (14) | C11—C10—C9 | 116.99 (14) |
| N3—C5—C6 | 106.05 (12) | C11—C10—H10 | 121.5 |
| C7—C6—C11 | 121.59 (13) | C9—C10—H10 | 121.5 |
| C7—C6—C5 | 130.30 (14) | C10—C11—C6 | 121.64 (13) |
| C11—C6—C5 | 108.10 (12) | C10—C11—C4 | 130.52 (13) |
| C6—C7—C8 | 116.96 (14) | C6—C11—C4 | 107.80 (12) |
| C6—C7—H7 | 121.5 | | |
| | | |
| C5—N3—C4—O1 | 178.06 (13) | C8—C9—C10—C11 | 0.1 (2) |
| C5—N3—C4—C11 | −0.65 (15) | C9—C10—C11—C6 | −0.3 (2) |
| C4—N3—C5—O2 | −179.86 (13) | C9—C10—C11—C4 | −177.73 (14) |
| C4—N3—C5—C6 | 0.17 (15) | C7—C6—C11—C10 | 0.2 (2) |
| O2—C5—C6—C7 | 1.6 (2) | C5—C6—C11—C10 | −178.76 (12) |
| N3—C5—C6—C7 | −178.46 (14) | C7—C6—C11—C4 | 178.20 (12) |
| O2—C5—C6—C11 | −179.55 (14) | C5—C6—C11—C4 | −0.81 (15) |
| N3—C5—C6—C11 | 0.42 (15) | O1—C4—C11—C10 | 0.0 (3) |
| C11—C6—C7—C8 | 0.0 (2) | N3—C4—C11—C10 | 178.61 (14) |
| C5—C6—C7—C8 | 178.78 (13) | O1—C4—C11—C6 | −177.74 (13) |
| C6—C7—C8—C9 | −0.2 (2) | N3—C4—C11—C6 | 0.90 (15) |
| C7—C8—C9—C10 | 0.2 (2) | | |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N3—H3···O2i | 0.88 | 2.02 | 2.8781 (16) | 167 |
| C7—H7···O2ii | 0.95 | 2.54 | 3.3874 (18) | 149 |
| C8—H8···O1iii | 0.95 | 2.56 | 3.4628 (19) | 159 |
| C10—H10···O1iv | 0.95 | 2.55 | 3.2303 (18) | 129 |
| Symmetry codes: (i) −x+1, −y+2, −z; (ii) −x, −y+1, −z; (iii) x−1, y−1, z; (iv) −x+3/2, y−1/2, −z+1/2. |
Experimental details
| Crystal data |
| Chemical formula | C8H5NO2 |
| Mr | 147.13 |
| Crystal system, space group | Monoclinic, P21/n |
| Temperature (K) | 150 |
| a, b, c (Å) | 3.7303 (2), 7.6638 (4), 22.6435 (13) |
| β (°) | 90.369 (2) |
| V (Å3) | 647.33 (6) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.11 |
| Crystal size (mm) | 0.15 × 0.05 × 0.05 |
| |
| Data collection |
| Diffractometer | KappaCCD
diffractometer |
| Absorption correction | Multi-scan
(DENZO-SMN; Otwinowski & Minor, 1997) |
| Tmin, Tmax | 0.984, 0.995 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 4750, 1456, 1061 |
| Rint | 0.054 |
| (sin θ/λ)max (Å−1) | 0.649 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.043, 0.103, 1.02 |
| No. of reflections | 1456 |
| No. of parameters | 101 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.22, −0.22 |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N3—H3···O2i | 0.88 | 2.02 | 2.8781 (16) | 167 |
| C7—H7···O2ii | 0.95 | 2.54 | 3.3874 (18) | 149 |
| C8—H8···O1iii | 0.95 | 2.56 | 3.4628 (19) | 159 |
| C10—H10···O1iv | 0.95 | 2.55 | 3.2303 (18) | 129 |
| Symmetry codes: (i) −x+1, −y+2, −z; (ii) −x, −y+1, −z; (iii) x−1, y−1, z; (iv) −x+3/2, y−1/2, −z+1/2. |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](https://journals.iucr.org/c/issues/2002/01/00/sk1520/sk1520fig1thm.gif)
![[Figure 2]](https://journals.iucr.org/c/issues/2002/01/00/sk1520/sk1520fig2thm.gif)




The structure of phthalimide [1H-isoindole-1,3(2H)-dione, C8H5NO2, I] was first reported (Matzat, 1972) using data collected from a large crystal of the mineral kladnoite: the structure was refined to R = 0.094 using ambient-temperature data. This report defined a dimeric structure formed by paired N—H···O hydrogen bonds and it alluded to some fairly short (less than 3.6 Å) intermolecular C···O distances: however, since at the time of publication (1972) the notion of C—H···O hydrogen bonds was effectively under anathema (Desiraju & Steiner, 1999), no molecular aggregation beyond dimer formation was discussed. In a later investigation (Ng, 1992), the structure was refined to R = 0.038, also from ambient-temperature data, but this report made no mention of C—H···O hydrogen bonds.
Following our investigation of intermolecular aggregation via hydrogen bonding and π···π stacking interactions in N,N'-dithiodiphthalimide (Skakle et al., 2001), we have now re-investigated phthalimide itself at 120 (2) K, and we have re-refined the structure using a data set rather larger than that employed by Ng (1992), where refinement on F with 770 reflections labelled observed gave R = 0.038, Δ/σ = 0.01 and S = 0.54 for 120 variables (n/p = 6.4), including isotropic refinement of all H parameters: the present refinement uses 1456 reflections giving n/p of 14.4. The intramolecular bonded distances found here are slightly more precise than those reported earlier (Ng, 1992), but show no significant variations from them, and hence will not be discussed further. In addition to the dimeric units generated by the hard hydrogen bonds, as described earlier (Matzat, 1972; Ng, 1992), the structure also contains two significant C—H···O hydrogen bonds which link the dimeric units into perforated molecular ribbons of some complexity.
There are three hydrogen bonds, one of N—H···O type and two of C—H···O type (Table 1), each of which alone generates a specific motif. The N—H···O hydrogen bond, where N3 at (x, y, z) (Fig. 1) acts as donor to O2 at (1 - x, 2 - y, -z), generates a centrosymmetric R22(8) ring centred at (1/2, 1, 0); C7 at (x, y, z) similarly acts as hydrogen-bond donor to O2 at (-x, 1 - y, -z), generating a second R22(8) ring centred at (0, 1/2, 0). The combination and propagation of these two motifs thus generates a chain of fused rings running parallel to the [110] direction, in which the rings formed by the hard hydrogen bonds are centred at (n-0.5, n, 0) (n = zero or integer) and those formed by the soft hydrogen bonds are centred at (n, n + 1/2, 0) (Fig. 2). Finally C8 at (x, y, z) acts as hydrogen-bond donor to O1 at (-1 + x, -1 + y, z), generating by translation a C(7) chain which serves to reinforce the chain of fused rings by the addition of peripheral R23(9) rings (Fig. 2), so generating a broad ribbon pierced by three types of ring.
It is striking how closely the R23(9) ring observed in (I) resembles that observed in the structure of 1,4-benzoquinone, where this motif links molecular chains built from R22(8) rings into a sheet (Trotter, 1960; Thalladi et al., 1998); in (I) this motif links R22(8) dimers into a ribbon.
There are two ribbons passing through each unit cell in (I), one in the domain -0.24 < z < +0.24 (Fig. 2), and the other in the domain 0.26 < z < 0.74. The only direction-specific interaction between adjacent ribbons is a very weak C—H···O contact having a C—H···O angle of 129° (Table 1), probably too small to be structurally significant. There are no aromatic π···π stacking interactions in the crystal structure of (I). It is interesting to note that the three C—H···O interactions in Table 1 correspond to three of the four shortest intermolecular C···O contacts noted by Matzat (1972), although their significance was obscured, both by the historical context, and by the emphasis on C···O distances at the expense of H···O distances and C—H···O angles. The fourth of Matzat's short C···O contacts (Matzat, 1972) is between C9 in the molecule at (x, y, z) and O1 at (1.5 - x, -0.5 + y, 0.5 - z), but this has C···O 3.3722 (19) Å, associated with a very long H···O distance (2.82 Å) and a narrow C—H···O angle (118°).