Buy article online - an online subscription or single-article purchase is required to access this article.

A microcrystalline carboxyl-functionalized imidazolium chloride, namely 1-carboxymethyl-3-ethylimidazolium chloride, C
7H
11N
2O
2+·Cl
−, has been synthesized and characterized by elemental analysis, attenuated total reflectance Fourier transform IR spectroscopy (ATR-FT-IR), single-crystal X-ray diffraction, thermal analysis (TGA/DSC), and photoluminescence spectroscopy. In the crystal structure, cations and anions are linked by C—H

Cl and C—H
O hydrogen bonds to create a helix along the [010] direction. Adjacent helical chains are further interconnected through O—H
Cl and C—H
O hydrogen bonds to form a (10
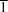
) layer. Finally, neighboring layers are joined together
via C—H
Cl contacts to generate a three-dimensional supramolecular architecture. Thermal analyses reveal that the compound melts at 449.7 K and is stable up to 560.0 K under a dynamic air atmosphere. Photoluminescence measurements show that the compound exhibits a blue fluorescence and a green phosphorescence associated with spin-allowed (
1π←
1π*) and spin-forbidden (
1π←
3π*) transitions, respectively. The average luminescence lifetime was determined to be 1.40 ns for the short-lived (
1π←
1π*) transition and 105 ms for the long-lived (
1π←
3π*) transition.
Supporting information
CCDC reference: 1559416
Data collection: SMART (Bruker, 2002); cell refinement: SAINT (Bruker, 2002); data reduction: SAINT (Bruker, 2002); program(s) used to solve structure: SHELXT (Sheldrick, 2015a); program(s) used to refine structure: SHELXL2014 (Sheldrick, 2015b); molecular graphics: DIAMOND (Brandenburg & Putz, 1999); software used to prepare material for publication: SHELXL2014 (Sheldrick, 2015b).
1-Carboxymethyl-3-ethylimidazolium chloride
top
Crystal data top
| C7H11N2O2+·Cl− | Dx = 1.347 Mg m−3 |
| Mr = 190.63 | Melting point: 446.1 K |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| a = 10.2225 (16) Å | Cell parameters from 4273 reflections |
| b = 9.6736 (16) Å | θ = 3.0–26.2° |
| c = 10.2678 (17) Å | µ = 0.37 mm−1 |
| β = 112.165 (8)° | T = 294 K |
| V = 940.3 (3) Å3 | Parallelepiped, colourless |
| Z = 4 | 0.10 × 0.10 × 0.10 mm |
| F(000) = 400 | |
Data collection top
Bruker APEX CCD
diffractometer | 1844 independent reflections |
| Radiation source: fine-focus sealed tube | 1557 reflections with I > 2σ(I) |
| Detector resolution: 8.33 pixels mm-1 | Rint = 0.031 |
| ω scans | θmax = 26.0°, θmin = 3.0° |
Absorption correction: empirical (using intensity measurements)
(Blessing, 1995) | h = −12→12 |
| Tmin = 0.911, Tmax = 0.983 | k = −11→11 |
| 12035 measured reflections | l = −12→12 |
Refinement top
| Refinement on F2 | 0 restraints |
| Least-squares matrix: full | Hydrogen site location: mixed |
| R[F2 > 2σ(F2)] = 0.036 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.106 | w = 1/[σ2(Fo2) + (0.0564P)2 + 0.1633P]
where P = (Fo2 + 2Fc2)/3 |
| S = 1.10 | (Δ/σ)max < 0.001 |
| 1844 reflections | Δρmax = 0.23 e Å−3 |
| 112 parameters | Δρmin = −0.20 e Å−3 |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| C1 | 0.13632 (17) | 0.67336 (15) | −0.00212 (17) | 0.0422 (4) | |
| H1 | 0.0773 | 0.7390 | 0.0133 | 0.051* | |
| C2 | 0.2695 (2) | 0.56244 (18) | −0.09152 (19) | 0.0528 (4) | |
| H2A | 0.3172 | 0.5395 | −0.1498 | 0.063* | |
| C3 | 0.2714 (2) | 0.49169 (18) | 0.0212 (2) | 0.0552 (5) | |
| H3 | 0.3207 | 0.4104 | 0.0560 | 0.066* | |
| C4 | 0.16677 (18) | 0.52539 (17) | 0.20420 (16) | 0.0448 (4) | |
| H4B | 0.1726 | 0.4258 | 0.2162 | 0.054* | |
| H4A | 0.0733 | 0.5543 | 0.1966 | 0.054* | |
| C5 | 0.27599 (17) | 0.59300 (16) | 0.33095 (16) | 0.0425 (4) | |
| C6 | 0.1495 (2) | 0.7779 (2) | −0.2195 (2) | 0.0571 (5) | |
| H6A | 0.2362 | 0.8134 | −0.2245 | 0.069* | |
| H6B | 0.0990 | 0.8548 | −0.1996 | 0.069* | |
| C7 | 0.0606 (2) | 0.7146 (3) | −0.3581 (2) | 0.0749 (6) | |
| H7A | 0.0397 | 0.7831 | −0.4308 | 0.112* | |
| H7B | 0.1111 | 0.6394 | −0.3785 | 0.112* | |
| H7C | −0.0259 | 0.6808 | −0.3537 | 0.112* | |
| N1 | 0.18375 (14) | 0.67547 (12) | −0.10510 (14) | 0.0414 (3) | |
| N2 | 0.18709 (14) | 0.56148 (13) | 0.07581 (13) | 0.0405 (3) | |
| O1 | 0.35277 (13) | 0.68432 (12) | 0.32434 (13) | 0.0516 (3) | |
| O2 | 0.27241 (16) | 0.54072 (15) | 0.44750 (13) | 0.0652 (4) | |
| H2 | 0.338 (3) | 0.587 (3) | 0.528 (3) | 0.098* | |
| Cl1 | 0.53307 (5) | 0.32422 (5) | 0.29617 (5) | 0.05769 (19) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| C1 | 0.0474 (9) | 0.0362 (8) | 0.0458 (9) | 0.0023 (6) | 0.0206 (7) | 0.0022 (7) |
| C2 | 0.0682 (11) | 0.0491 (10) | 0.0482 (10) | 0.0122 (8) | 0.0299 (9) | 0.0008 (8) |
| C3 | 0.0733 (12) | 0.0441 (9) | 0.0516 (11) | 0.0180 (8) | 0.0276 (9) | 0.0050 (8) |
| C4 | 0.0548 (9) | 0.0410 (8) | 0.0400 (9) | −0.0083 (7) | 0.0195 (8) | 0.0006 (7) |
| C5 | 0.0521 (9) | 0.0390 (8) | 0.0384 (9) | −0.0022 (7) | 0.0192 (8) | 0.0015 (6) |
| C6 | 0.0629 (11) | 0.0543 (10) | 0.0619 (12) | 0.0082 (8) | 0.0322 (10) | 0.0213 (9) |
| C7 | 0.0759 (14) | 0.1035 (17) | 0.0478 (12) | 0.0119 (12) | 0.0260 (11) | 0.0248 (11) |
| N1 | 0.0469 (7) | 0.0385 (7) | 0.0402 (8) | 0.0017 (5) | 0.0179 (6) | 0.0032 (5) |
| N2 | 0.0487 (7) | 0.0365 (7) | 0.0349 (7) | −0.0013 (5) | 0.0142 (6) | −0.0012 (5) |
| O1 | 0.0616 (7) | 0.0491 (7) | 0.0443 (7) | −0.0149 (5) | 0.0201 (6) | −0.0002 (5) |
| O2 | 0.0866 (10) | 0.0714 (9) | 0.0375 (7) | −0.0274 (7) | 0.0232 (7) | 0.0024 (6) |
| Cl1 | 0.0604 (3) | 0.0721 (3) | 0.0408 (3) | −0.0079 (2) | 0.0194 (2) | 0.0024 (2) |
Geometric parameters (Å, º) top
| C1—N1 | 1.319 (2) | C4—H4A | 0.9700 |
| C1—N2 | 1.330 (2) | C5—O1 | 1.2005 (19) |
| C1—H1 | 0.9300 | C5—O2 | 1.3124 (19) |
| C2—C3 | 1.339 (3) | C6—N1 | 1.474 (2) |
| C2—N1 | 1.375 (2) | C6—C7 | 1.501 (3) |
| C2—H2A | 0.9300 | C6—H6A | 0.9700 |
| C3—N2 | 1.370 (2) | C6—H6B | 0.9700 |
| C3—H3 | 0.9300 | C7—H7A | 0.9600 |
| C4—N2 | 1.4524 (19) | C7—H7B | 0.9600 |
| C4—C5 | 1.507 (2) | C7—H7C | 0.9600 |
| C4—H4B | 0.9700 | O2—H2 | 0.96 (3) |
| | | |
| N1—C1—N2 | 108.48 (13) | N1—C6—H6A | 109.4 |
| N1—C1—H1 | 125.8 | C7—C6—H6A | 109.4 |
| N2—C1—H1 | 125.8 | N1—C6—H6B | 109.4 |
| C3—C2—N1 | 107.16 (15) | C7—C6—H6B | 109.4 |
| C3—C2—H2A | 126.4 | H6A—C6—H6B | 108.0 |
| N1—C2—H2A | 126.4 | C6—C7—H7A | 109.5 |
| C2—C3—N2 | 107.19 (15) | C6—C7—H7B | 109.5 |
| C2—C3—H3 | 126.4 | H7A—C7—H7B | 109.5 |
| N2—C3—H3 | 126.4 | C6—C7—H7C | 109.5 |
| N2—C4—C5 | 111.49 (13) | H7A—C7—H7C | 109.5 |
| N2—C4—H4B | 109.3 | H7B—C7—H7C | 109.5 |
| C5—C4—H4B | 109.3 | C1—N1—C2 | 108.62 (13) |
| N2—C4—H4A | 109.3 | C1—N1—C6 | 126.88 (14) |
| C5—C4—H4A | 109.3 | C2—N1—C6 | 124.47 (14) |
| H4B—C4—H4A | 108.0 | C1—N2—C3 | 108.54 (14) |
| O1—C5—O2 | 125.37 (16) | C1—N2—C4 | 125.80 (13) |
| O1—C5—C4 | 123.90 (14) | C3—N2—C4 | 125.46 (14) |
| O2—C5—C4 | 110.71 (14) | C5—O2—H2 | 111.1 (16) |
| N1—C6—C7 | 110.98 (16) | | |
| | | |
| N1—C2—C3—N2 | 0.0 (2) | C7—C6—N1—C2 | 67.4 (2) |
| N2—C4—C5—O1 | 12.9 (2) | N1—C1—N2—C3 | 0.97 (19) |
| N2—C4—C5—O2 | −168.70 (14) | N1—C1—N2—C4 | 176.09 (14) |
| N2—C1—N1—C2 | −0.95 (19) | C2—C3—N2—C1 | −0.6 (2) |
| N2—C1—N1—C6 | 177.42 (15) | C2—C3—N2—C4 | −175.75 (15) |
| C3—C2—N1—C1 | 0.6 (2) | C5—C4—N2—C1 | −85.89 (19) |
| C3—C2—N1—C6 | −177.86 (17) | C5—C4—N2—C3 | 88.4 (2) |
| C7—C6—N1—C1 | −110.8 (2) | | |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C3—H3···Cl1 | 0.93 | 2.73 | 3.472 (2) | 137 |
| C1—H1···Cl1i | 0.93 | 2.73 | 3.5161 (19) | 143 |
| C4—H4B···O1ii | 0.97 | 2.37 | 3.312 (2) | 164 |
| O2—H2···Cl1iii | 0.96 (3) | 1.98 (3) | 2.9393 (15) | 176 (2) |
| C1—H1···O1iv | 0.93 | 2.49 | 3.094 (2) | 122 |
| C2—H2A···Cl1v | 0.93 | 2.84 | 3.590 (2) | 138 |
| Symmetry codes: (i) −x+1/2, y+1/2, −z+1/2; (ii) −x+1/2, y−1/2, −z+1/2; (iii) −x+1, −y+1, −z+1; (iv) x−1/2, −y+3/2, z−1/2; (v) −x+1, −y+1, −z. |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu research papers
research papers













 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry


