Buy article online - an online subscription or single-article purchase is required to access this article.
In the title molecular complex, [Cu
4Cl
6O(2-EtTz)
4], where 2-EtTz is 2-ethyltetrazole (C
3H
6N
4), the central O atom is located on the
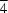
symmetry site and is tetrahedrally coordinated to four Cu atoms, with Cu—O distances of 1.8966 (4) Å. A very slight distortion of Cu
4O from a regular tetrahedron is observed [two Cu—O—Cu angles are 108.76 (3)° and four others are 109.828 (13)°]. Each Cu atom is connected to three others
via the Cl atoms, forming a slightly distorted Cl octahedron around the O atom, with O

Cl distances of 2.9265 (7) Å for Cl atoms lying on the twofold axis and 2.9441 (13) Å for those in general positions. The Cu atom has a distorted trigonal–bipyramidal environment, with three Cl atoms in the equatorial plane, and with the N atom of the 2-ethyltetrazole ligand and the μ
4-O atom in axial positions. The Cu atom is displaced out of the equatorial plane by
ca 0.91 Å towards the coordinated N atom of the 2-ethyltetrazole ligand.
Supporting information
CCDC reference: 248136
2-Ethyltetrazole (1.57 g, 0.016 mol) was added to a solution of CuCl2·2H2O (2.82 g, 0.0165 mol) in methanol (20 ml). The reaction mixture was stirred for 1 h and was then kept in air at room temperature. Two months later, a mixture of needle-like green and prismatic brown crystals had formed, from which prismatic brown crystals of (I) (0.52 g, yield ca 15%) could be separated. Analysis found: Cu 29.1, Cl 23.1%; calculated: Cu 29.0, Cl 24.3%. These values correspond to the composition [Cu4OCl6(2-EtTz)4].
The H atoms were placed in geometrically calculated positions, with C—H distances in the range 0.93–0.97 Å, and refined using a riding model, with Uiso(H) = 1.5Ueq(C) for the methyl group and 1.2Ueq(C) for the other H atoms.
Data collection: R3m Software (Nicolet, 1980); cell refinement: R3m Software; data reduction: R3m Software; program(s) used to solve structure: SIR97 (Altomare et al., 1999); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEP-3 for Windows (Farrugia, 1997); software used to prepare material for publication: SHELXL97.
Hexa-µ
2-chloro-µ
4-oxo-tetrakis[(2-ethyltetrazole-kN
4)copper(II)]
top
Crystal data top
| [Cu4Cl6O(C3H6N4)4] | Dx = 1.902 Mg m−3 |
| Mr = 875.37 | Mo Kα radiation, λ = 0.71073 Å |
| Tetragonal, I41/a | Cell parameters from 25 reflections |
| Hall symbol: -I 4ad | θ = 18.1–21.4° |
| a = 16.069 (2) Å | µ = 3.31 mm−1 |
| c = 11.836 (4) Å | T = 293 K |
| V = 3056.2 (12) Å3 | Prism, dark brown |
| Z = 4 | 0.40 × 0.38 × 0.35 mm |
| F(000) = 1736 | |
Data collection top
Nicolet R3m four-circle
diffractometer | 1938 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.025 |
| Graphite monochromator | θmax = 30.0°, θmin = 2.1° |
| ω/2θ scans | h = 0→22 |
Absorption correction: multi-scan
(Blessing, 1995) | k = 0→22 |
| Tmin = 0.281, Tmax = 0.312 | l = −1→16 |
| 2607 measured reflections | 3 standard reflections every 100 reflections |
| 2243 independent reflections | intensity decay: none |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.034 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.093 | H-atom parameters constrained |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.048P)2 + 3.5779P]
where P = (Fo2 + 2Fc2)/3 |
| 2243 reflections | (Δ/σ)max = 0.001 |
| 90 parameters | Δρmax = 0.64 e Å−3 |
| 0 restraints | Δρmin = −0.55 e Å−3 |
Crystal data top
| [Cu4Cl6O(C3H6N4)4] | Z = 4 |
| Mr = 875.37 | Mo Kα radiation |
| Tetragonal, I41/a | µ = 3.31 mm−1 |
| a = 16.069 (2) Å | T = 293 K |
| c = 11.836 (4) Å | 0.40 × 0.38 × 0.35 mm |
| V = 3056.2 (12) Å3 | |
Data collection top
Nicolet R3m four-circle
diffractometer | 1938 reflections with I > 2σ(I) |
Absorption correction: multi-scan
(Blessing, 1995) | Rint = 0.025 |
| Tmin = 0.281, Tmax = 0.312 | 3 standard reflections every 100 reflections |
| 2607 measured reflections | intensity decay: none |
| 2243 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.034 | 0 restraints |
| wR(F2) = 0.093 | H-atom parameters constrained |
| S = 1.05 | Δρmax = 0.64 e Å−3 |
| 2243 reflections | Δρmin = −0.55 e Å−3 |
| 90 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Cu | 0.944202 (17) | 0.171949 (17) | 0.71832 (2) | 0.03234 (10) | |
| Cl1 | 1.0000 | 0.2500 | 0.87374 (7) | 0.0488 (2) | |
| Cl2 | 0.82293 (4) | 0.20827 (5) | 0.61351 (7) | 0.0538 (2) | |
| O | 1.0000 | 0.2500 | 0.6250 | 0.0277 (6) | |
| N1 | 0.78912 (15) | 0.03369 (15) | 0.92310 (19) | 0.0442 (5) | |
| N2 | 0.86176 (13) | −0.00571 (13) | 0.92655 (17) | 0.0364 (4) | |
| N3 | 0.92013 (13) | 0.02813 (13) | 0.86534 (19) | 0.0393 (4) | |
| N4 | 0.88463 (13) | 0.09373 (12) | 0.81803 (17) | 0.0348 (4) | |
| C5 | 0.80601 (17) | 0.09580 (16) | 0.8540 (2) | 0.0411 (5) | |
| H5 | 0.7677 | 0.1362 | 0.8328 | 0.049* | |
| C6 | 0.8748 (2) | −0.08318 (17) | 0.9913 (2) | 0.0472 (6) | |
| H6A | 0.8387 | −0.0832 | 1.0569 | 0.057* | |
| H6B | 0.9319 | −0.0851 | 1.0178 | 0.057* | |
| C7 | 0.8575 (2) | −0.15846 (19) | 0.9218 (3) | 0.0610 (8) | |
| H7A | 0.7998 | −0.1590 | 0.9004 | 0.092* | |
| H7B | 0.8698 | −0.2075 | 0.9650 | 0.092* | |
| H7C | 0.8915 | −0.1575 | 0.8551 | 0.092* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Cu | 0.03468 (16) | 0.02963 (15) | 0.03271 (16) | −0.00347 (10) | 0.00210 (10) | 0.00381 (10) |
| Cl1 | 0.0675 (6) | 0.0499 (5) | 0.0291 (4) | −0.0169 (4) | 0.000 | 0.000 |
| Cl2 | 0.0320 (3) | 0.0663 (4) | 0.0630 (4) | −0.0104 (3) | −0.0079 (3) | 0.0285 (3) |
| O | 0.0274 (8) | 0.0274 (8) | 0.0283 (14) | 0.000 | 0.000 | 0.000 |
| N1 | 0.0452 (12) | 0.0475 (12) | 0.0399 (11) | 0.0054 (9) | 0.0133 (9) | 0.0085 (9) |
| N2 | 0.0420 (11) | 0.0349 (10) | 0.0322 (9) | −0.0018 (8) | 0.0033 (8) | 0.0049 (8) |
| N3 | 0.0375 (10) | 0.0359 (10) | 0.0445 (11) | −0.0011 (8) | 0.0018 (8) | 0.0084 (8) |
| N4 | 0.0395 (10) | 0.0307 (9) | 0.0341 (9) | −0.0021 (7) | 0.0029 (8) | 0.0028 (7) |
| C5 | 0.0446 (13) | 0.0395 (12) | 0.0392 (12) | 0.0080 (10) | 0.0114 (10) | 0.0037 (10) |
| C6 | 0.0575 (16) | 0.0410 (13) | 0.0431 (13) | −0.0033 (11) | −0.0049 (12) | 0.0139 (11) |
| C7 | 0.073 (2) | 0.0393 (14) | 0.071 (2) | −0.0043 (14) | −0.0159 (17) | 0.0096 (14) |
Geometric parameters (Å, º) top
| Cu—O | 1.8966 (4) | N2—N3 | 1.304 (3) |
| Cu—N4 | 1.972 (2) | N2—C6 | 1.477 (3) |
| Cu—Cl2 | 2.3827 (8) | N3—N4 | 1.323 (3) |
| Cu—Cl1 | 2.4002 (9) | N4—C5 | 1.333 (3) |
| Cu—Cl2i | 2.4344 (7) | C5—H5 | 0.9300 |
| Cl1—Cuii | 2.4002 (9) | C6—C7 | 1.489 (4) |
| Cl2—Cuiii | 2.4344 (7) | C6—H6A | 0.9700 |
| O—Cuii | 1.8966 (4) | C6—H6B | 0.9700 |
| O—Cui | 1.8966 (4) | C7—H7A | 0.9600 |
| O—Cuiii | 1.8966 (4) | C7—H7B | 0.9600 |
| N1—C5 | 1.319 (3) | C7—H7C | 0.9600 |
| N1—N2 | 1.329 (3) | | |
| | | |
| O—Cu—N4 | 178.20 (6) | N1—N2—C6 | 122.9 (2) |
| O—Cu—Cl2 | 85.50 (2) | N2—N3—N4 | 104.90 (19) |
| N4—Cu—Cl2 | 94.05 (6) | N3—N4—C5 | 107.1 (2) |
| O—Cu—Cl1 | 85.65 (3) | N3—N4—Cu | 123.49 (16) |
| N4—Cu—Cl1 | 93.20 (6) | C5—N4—Cu | 129.41 (17) |
| Cl2—Cu—Cl1 | 125.21 (3) | N1—C5—N4 | 112.0 (2) |
| O—Cu—Cl2i | 84.04 (2) | N1—C5—H5 | 124.0 |
| N4—Cu—Cl2i | 97.70 (6) | N4—C5—H5 | 124.0 |
| Cl2—Cu—Cl2i | 118.652 (19) | N2—C6—C7 | 111.8 (2) |
| Cl1—Cu—Cl2i | 113.95 (3) | N2—C6—H6A | 109.3 |
| Cu—Cl1—Cuii | 79.93 (4) | C7—C6—H6A | 109.3 |
| Cu—Cl2—Cuiii | 80.23 (2) | N2—C6—H6B | 109.3 |
| Cuii—O—Cu | 108.76 (3) | C7—C6—H6B | 109.3 |
| Cuii—O—Cui | 109.828 (13) | H6A—C6—H6B | 107.9 |
| Cu—O—Cui | 109.828 (13) | C6—C7—H7A | 109.5 |
| Cuii—O—Cuiii | 109.827 (13) | C6—C7—H7B | 109.5 |
| Cu—O—Cuiii | 109.827 (13) | H7A—C7—H7B | 109.5 |
| Cui—O—Cuiii | 108.76 (3) | C6—C7—H7C | 109.5 |
| C5—N1—N2 | 101.5 (2) | H7A—C7—H7C | 109.5 |
| N3—N2—N1 | 114.6 (2) | H7B—C7—H7C | 109.5 |
| N3—N2—C6 | 122.5 (2) | | |
| | | |
| O—Cu—Cl1—Cuii | 0.0 | C5—N1—N2—N3 | 0.1 (3) |
| N4—Cu—Cl1—Cuii | −178.61 (6) | C5—N1—N2—C6 | −177.9 (2) |
| Cl2—Cu—Cl1—Cuii | −81.37 (3) | N1—N2—N3—N4 | −0.1 (3) |
| Cl2i—Cu—Cl1—Cuii | 81.51 (2) | C6—N2—N3—N4 | 177.9 (2) |
| O—Cu—Cl2—Cuiii | −4.205 (18) | N2—N3—N4—C5 | 0.1 (3) |
| N4—Cu—Cl2—Cuiii | 174.05 (6) | N2—N3—N4—Cu | 178.20 (16) |
| Cl1—Cu—Cl2—Cuiii | 77.25 (4) | Cl2—Cu—N4—N3 | 151.24 (19) |
| Cl2i—Cu—Cl2—Cuiii | −84.90 (3) | Cl1—Cu—N4—N3 | −83.10 (19) |
| Cl2—Cu—O—Cuii | 125.87 (3) | Cl2i—Cu—N4—N3 | 31.6 (2) |
| Cl1—Cu—O—Cuii | 0.0 | Cl2—Cu—N4—C5 | −31.1 (2) |
| Cl2i—Cu—O—Cuii | −114.67 (3) | Cl1—Cu—N4—C5 | 94.5 (2) |
| Cl2—Cu—O—Cui | −113.91 (3) | Cl2i—Cu—N4—C5 | −150.8 (2) |
| Cl1—Cu—O—Cui | 120.217 (8) | N2—N1—C5—N4 | 0.0 (3) |
| Cl2i—Cu—O—Cui | 5.55 (2) | N3—N4—C5—N1 | 0.0 (3) |
| Cl2—Cu—O—Cuiii | 5.66 (2) | Cu—N4—C5—N1 | −177.98 (18) |
| Cl1—Cu—O—Cuiii | −120.217 (8) | N3—N2—C6—C7 | −87.6 (3) |
| Cl2i—Cu—O—Cuiii | 125.12 (3) | N1—N2—C6—C7 | 90.3 (3) |
| Symmetry codes: (i) −y+5/4, x−3/4, −z+5/4; (ii) −x+2, −y+1/2, z; (iii) y+3/4, −x+5/4, −z+5/4. |
Experimental details
| Crystal data |
| Chemical formula | [Cu4Cl6O(C3H6N4)4] |
| Mr | 875.37 |
| Crystal system, space group | Tetragonal, I41/a |
| Temperature (K) | 293 |
| a, c (Å) | 16.069 (2), 11.836 (4) |
| V (Å3) | 3056.2 (12) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 3.31 |
| Crystal size (mm) | 0.40 × 0.38 × 0.35 |
| |
| Data collection |
| Diffractometer | Nicolet R3m four-circle
diffractometer |
| Absorption correction | Multi-scan
(Blessing, 1995) |
| Tmin, Tmax | 0.281, 0.312 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 2607, 2243, 1938 |
| Rint | 0.025 |
| (sin θ/λ)max (Å−1) | 0.704 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.034, 0.093, 1.05 |
| No. of reflections | 2243 |
| No. of parameters | 90 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.64, −0.55 |
Selected geometric parameters (Å, º) top| Cu—O | 1.8966 (4) | Cu—Cl1 | 2.4002 (9) |
| Cu—N4 | 1.972 (2) | Cu—Cl2i | 2.4344 (7) |
| Cu—Cl2 | 2.3827 (8) | | |
| | | |
| O—Cu—N4 | 178.20 (6) | N4—Cu—Cl2i | 97.70 (6) |
| O—Cu—Cl2 | 85.50 (2) | Cl2—Cu—Cl2i | 118.652 (19) |
| N4—Cu—Cl2 | 94.05 (6) | Cl1—Cu—Cl2i | 113.95 (3) |
| O—Cu—Cl1 | 85.65 (3) | Cu—Cl1—Cuii | 79.93 (4) |
| N4—Cu—Cl1 | 93.20 (6) | Cu—Cl2—Cuiii | 80.23 (2) |
| Cl2—Cu—Cl1 | 125.21 (3) | Cuii—O—Cu | 108.76 (3) |
| O—Cu—Cl2i | 84.04 (2) | Cuii—O—Cui | 109.828 (13) |
| Symmetry codes: (i) −y+5/4, x−3/4, −z+5/4; (ii) −x+2, −y+1/2, z; (iii) y+3/4, −x+5/4, −z+5/4. |
Tetrazole ring bond lengths (Å) in (I) compared with the corresponding mean values (s.u. values in parentheses) for five metal(II) complexes of 2-substituted tetrazoles found in a CSD survey. top| Bond | (I) | Mean from CSD |
| N1-N2 | 1.329 (3) | 1.322 (2) |
| N1═C5 | 1.319 (3) | 1.319 (10) |
| N2-N3 | 1.304 (3) | 1.303 (3) |
| N3═N4 | 1.323 (3) | 1.323 (2) |
| N4-C5 | 1.333 (3) | 1.334 (5) |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu metal-organic compounds
metal-organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2004/08/00/jz1637/jz1637fig1thm.gif)

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry



Tetranuclear complexes of copper(II) with the composition Cu4OX6L4 (where X is Cl or Br, and L is Cl, Br or ligands containing N, O or P donors) have been the subject of numerous investigations, mainly because of their unusual magnetic properties, which are caused by the existence of two different exchange interaction channels, Cu—O—Cu and Cu—X—Cu, and which depend on the nature of the ligand L (Carlin, 1986; Atria et al., 1999). The high thermodynamic stability of such complexes is reflected in their frequent formation as by-products on preparing CuX2.nL adducts starting from the corresponding copper(II) halide hydrate and compound L (Norman & Rose, 1989; Virovets et al., 2001).
We have recently shown that Cu4OX6L4 complexes are also formed in the attempt to synthesize copper(II) chloride complexes with 2-substituted tetazoles, especially on slow crystallization (Degtyarik et al., 2003). Three such complexes, with L = 2-methyl-, 2-ethyl- and 2-allyltetrazole, have been obtained. This paper presents the crystal structure of one of them, [Cu4OCl6(2-EtTz)4], (I), where 2-EtTz is 2-ethyltetrazole (Fig. 1). \sch
The molecule of (I) consists of a tetrahedron of Cu atoms bonded to a central O atom. Each Cu atom is connected to three others through bridging Cl atoms, and to the 2-ethyltetrazole ligand via atom N4. The O and Cl1 atoms lie on Wyckoff sites 4 and 2, respectively; all others are in general positions.
The crystallographic site symmetry of the central O atom imposes 4 symmetry on the Cu4O tetrahedron and of course on the molecule as a whole. The tetrahedron has Cu—O bond lengths of 1.8966 (4) Å, and two Cu—O—Cu angles of 108.76 (3) Å (the 4 axis lies on the intersection of the planes of these angles) and four others of 109.828 (13) Å. The Cu···Cu distances corresponding to the above Cu—O—Cu angles are 3.1039 (7) and 3.0835 (7) Å, respectively.
Six Cl atoms of the molecule form a slightly distorted octahedron around the O atom, with two axial O···Cl1 distances of 2.9441 (13) Å and four equatorial O···Cl2 distances of 2.9265 (7) Å. The Cl2···O···Cl2 equatorial-equatorial angles are either 90.124 (2) or 174.68 (3)°. The Cl1···O···Cl2 axial-equatorial angles are 92.662 (16) or 87.338 (16)°.
The Cu atom has a distorted trigonal-bipyramidal coordination (Table 1), with a τ descriptor of 0.88 (extreme values are 0 for a square pyramid and 1 for a trigonal bipyramid; Addison et al., 1984). The axial positions of the bipyramid are occupied by the O atom and atom N4 of one 2-ethyltetrazole ligand, with Cu—O and Cu—N4 bond lengths of 1.8966 (4) and 1.972 (2) Å, respectively. The equatorial plane is occupied by one Cl1 atom [Cu—Cl1 2.4002 (9) Å] and two Cl2 atoms [Cu—Cl2 2.3827 (8) and 2.4344 (7) Å]. The axial-axial O—Cu—N4 angle of the bipyramid deviates slightly from 180°, with a value of 178.20 (6)°. The Cl2—Cu—Cl2 and Cl2—Cu—Cl1 equatorial-equatorial angles are 118.652 (19) and 113.95 (3)°, respectively. The axial-equatorial angles fall into two sets, with O—Cu—Cl values in the range 84.04 (2)–85.65 (3)° and N4—Cu—Cl in the range 93.20 (6)–97.70 (6)°, which reflects the fact that the Cu atom is displaced out of the plane of the three equatorial Cl atoms by ca 0.91 Å towards atom N4 of the 2-ethyltetrazole ligand. The Cu—Cl1—Cu and Cu—Cl2—Cu bridges are characterized by angles of 79.93 (4) and 80.23 (3)°, respectively.
The tetrazole ring of the 2-ethyltetrazole ligand in (I) is essentially planar, with a mean deviation from the least-squares plane of 0.0005 (16) Å. The tetrazole ring geometry is similar to those found previously for complexes of 2-substituted tetrazoles, namely [Ni(2-MeTz)6](BF4)2 (MeTz = 2-methyltetrazole; van den Heuvel et al., 1983), [ZnL3](ClO4)2 (L = 1,2-bis(tetrazol-2-yl)ethane; Bronisz, 2002), [CuCl2(2-tBuTz)] (2-tBuTz = 2-tert-buthyltetrazole; Lyakhov Gaponik Degtyarik & Ivashkevich, 2003a), [CuCl3(2-AlTz)4] (2-AlTz = 2-allyltetrazole; Lyakhov Gaponik Degtyarik et al., 2003) and [CuCl2(2-EtTz)2] (2-EtTz = 2-ethyltetrazole; (Lyakhov Gaponik Degtyarik & Ivashkevich, 2003b). Table 2 compares the tetrazole ring bond lengths in (I) with those for these? previously investigated complexes of 2-substituted tetrazoles found in an analysis of search results from the Cambridge Structural Database (CSD, Version 5.24, November 2002 release; Allen 2002).
Because of lack of hydrogen bonds in the structure of (I), only van der Waals interactions are responsible for the crystal packing.