

 journal menu
journal menuBuy article online - an online subscription or single-article purchase is required to access this article.
 research papers
research papers









The crystal structure of the 224-residue protein dethiobiotin synthetase from Escherichia coli has been refined using X-ray diffraction data at 0.97 Å resolution at 100 K. The model, consisting of 4143 protein atoms including 1859 H atoms and 436 solvent sites, was refined to a final R factor of 11.6% for all reflections, and has an estimated mean standard uncertainty for the atomic positions of 0.022 Å, derived from inversion of the blocked matrix. The structure was refined with a full anisotropic model for the atomic displacement parameters using SHELX97. Stereochemical restraints were applied throughout the refinement. In the last cycles, the planarity of the peptide bonds was not restrained, resulting in a mean ω value of 179.6°. Analysis of the most anisotropic regions of the protein shows that they form four clusters of residues. Alternate conformations for the side chains of 15 residues and for the main-chain atoms of six residues from three loops were included in the model. An analysis of C—H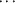 O hydrogen bonds shows that such interactions occur rather frequently in DTBS; in total, 16 such hydrogen bonds were found. In the central β-sheet, 13 C—HO bonds between carbonyl O and Cα H atoms were found. Other interactions of this type involve main-chain–side-chain and side-chain–side-chain C—HO bonds. The model includes 436 water sites, of which 233 molecules form the first hydration shell. Analysis of the protein–solvent interactions shows that about one third of the accessible surface of the enzyme is not covered by ordered solvent. No difference in propensity for ordered solvent close to hydrophilic or hydrophobic surface areas was found. The comparison of the 100 K structure with the structure of the enzyme determined at room temperature shows several regions with different conformation, including areas in the active site, suggesting that structural transitions can occur during flash freezing. This observation questions one of the basic assumptions in the analysis of enzymatic reaction mechanisms using cryocrystallography.
O hydrogen bonds shows that such interactions occur rather frequently in DTBS; in total, 16 such hydrogen bonds were found. In the central β-sheet, 13 C—HO bonds between carbonyl O and Cα H atoms were found. Other interactions of this type involve main-chain–side-chain and side-chain–side-chain C—HO bonds. The model includes 436 water sites, of which 233 molecules form the first hydration shell. Analysis of the protein–solvent interactions shows that about one third of the accessible surface of the enzyme is not covered by ordered solvent. No difference in propensity for ordered solvent close to hydrophilic or hydrophobic surface areas was found. The comparison of the 100 K structure with the structure of the enzyme determined at room temperature shows several regions with different conformation, including areas in the active site, suggesting that structural transitions can occur during flash freezing. This observation questions one of the basic assumptions in the analysis of enzymatic reaction mechanisms using cryocrystallography.

 Subscribe to Acta Crystallographica Section D: Biological Crystallography
Subscribe to Acta Crystallographica Section D: Biological Crystallography


