Buy article online - an online subscription or single-article purchase is required to access this article.
In the structure of the title compound, C
6H
18N
22+·H(C
2H
2ClO
2)
2−·Cl
−, the hexane-1,6-diaminium dication is disordered over two sets of positions, with almost equal occupancies. Both alternative positions of the dication are in the fully extended conformation, situated on an inversion centre at (
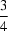
,
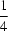
,
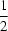
). Two chloroacetic acid moieties, related by another centre of symmetry at (
,
,
), are connected by a very short symmetrical O

H
O hydrogen bond [O
O = 2.452 (2) Å], with the H atom at the centre of inversion. These two fragments thus effectively form the hydrogen bis(chloroacetate) monoanion, and the overall charge is balanced by an additional chloride anion which resides on a twofold axis. The ions form a layer structure, with alternating layers of dications and anions running along the [101] direction, linked
via hydrogen bonds. There are two N—H
O interactions and two N—H
Cl
− interactions.
Supporting information
CCDC reference: 760126
1,6-Diaminohexane (1 mmol) was dissolved in water–methanol (1:1) (2 ml) and
mixed successively with chloroacetic acid and hydrochloric acid (2:1) (1.4 mmol). The excess of acid was used to assure complete protonation of both
amine groups. The solution was heated and placed at ambient temperature for
slow crystalization. After a few days, transparent crystals suitable for X-ray
diffraction were obtained.
The disorder applies to all hexane-1,6-diaminium atoms; two forms of analogous
dications cross each other at the middle of C4—C4' bond, at the inversion
center. All H atoms of both forms of the dication were positioned
geometrically (N—H = 0.89Å and C—H = 0.97Å) and were refined using a
riding model, with Uiso values set at 1.2 (CH2 groups) and 1.5
(NH3) times the Ueq value of the carrier atom. C—N bond lengths
were restrained to a target value of 1.49 (1)Å.
Data collection: CrysAlis PRO (Oxford Diffraction, 2009\); cell refinement: CrysAlis PRO (Oxford Diffraction, 2009\); data reduction: CrysAlis PRO (Oxford Diffraction, 2009\); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008\); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008\); molecular graphics: Stereochemical Workstation Operation Manual (Siemens, 1989\)
and
Mercury (Macrae et al., 2008\); software used to prepare material for publication: SHELXL97 (Sheldrick, 2008\).
hexane-1,6-diaminium chloride chloroacetate - chloroacetic acid
top
Crystal data top
| C6H18N22+·C4H5Cl2O4−·Cl− | F(000) = 720 |
| Mr = 341.65 | Dx = 1.369 Mg m−3 |
| Monoclinic, C2/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -C 2yc | Cell parameters from 1639 reflections |
| a = 25.702 (5) Å | θ = 2.0–26.8° |
| b = 4.4419 (4) Å | µ = 0.56 mm−1 |
| c = 18.718 (4) Å | T = 100 K |
| β = 129.14 (3)° | Block, colourless |
| V = 1657.4 (9) Å3 | 0.3 × 0.3 × 0.2 mm |
| Z = 4 | |
Data collection top
Xcalibur, Eos
diffractometer | 1301 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.027 |
| Graphite monochromator | θmax = 26.7°, θmin = 4.1° |
| Detector resolution: 8.1929 pixels mm-1 | h = −31→23 |
| ω scan | k = −5→4 |
| 2850 measured reflections | l = −21→23 |
| 1614 independent reflections | |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.029 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.062 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.0281P)2]
where P = (Fo2 + 2Fc2)/3 |
| 1614 reflections | (Δ/σ)max = 0.001 |
| 135 parameters | Δρmax = 0.20 e Å−3 |
| 2 restraints | Δρmin = −0.33 e Å−3 |
Crystal data top
| C6H18N22+·C4H5Cl2O4−·Cl− | V = 1657.4 (9) Å3 |
| Mr = 341.65 | Z = 4 |
| Monoclinic, C2/c | Mo Kα radiation |
| a = 25.702 (5) Å | µ = 0.56 mm−1 |
| b = 4.4419 (4) Å | T = 100 K |
| c = 18.718 (4) Å | 0.3 × 0.3 × 0.2 mm |
| β = 129.14 (3)° | |
Data collection top
Xcalibur, Eos
diffractometer | 1301 reflections with I > 2σ(I) |
| 2850 measured reflections | Rint = 0.027 |
| 1614 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.029 | 2 restraints |
| wR(F2) = 0.062 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.05 | Δρmax = 0.20 e Å−3 |
| 1614 reflections | Δρmin = −0.33 e Å−3 |
| 135 parameters | |
Special details top
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell esds are taken
into account individually in the estimation of esds in distances, angles
and torsion angles; correlations between esds in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell esds is used for estimating esds involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and
goodness of fit S are based on F2, conventional R-factors R are based
on F, with F set to zero for negative F2. The threshold expression of
F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is
not relevant to the choice of reflections for refinement. R-factors based
on F2 are statistically about twice as large as those based on F, and R-
factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | Occ. (<1) |
| N1A | 0.5621 (3) | 0.3568 (16) | 0.2157 (4) | 0.0140 (11) | 0.488 (8) |
| H1A1 | 0.5451 | 0.1974 | 0.2234 | 0.021* | 0.488 (8) |
| H1A2 | 0.5492 | 0.3550 | 0.1590 | 0.021* | 0.488 (8) |
| H1A3 | 0.5475 | 0.5244 | 0.2240 | 0.021* | 0.488 (8) |
| N1B | 0.5761 (3) | 0.3635 (14) | 0.2074 (4) | 0.0126 (10) | 0.512 (8) |
| H1B1 | 0.5853 | 0.2084 | 0.1869 | 0.019* | 0.512 (8) |
| H1B2 | 0.5579 | 0.5113 | 0.1660 | 0.019* | 0.512 (8) |
| H1B3 | 0.5474 | 0.3056 | 0.2163 | 0.019* | 0.512 (8) |
| C2A | 0.6370 (3) | 0.3461 (14) | 0.2844 (4) | 0.0135 (10) | 0.488 (8) |
| H2A | 0.6553 | 0.5297 | 0.2802 | 0.016* | 0.488 (8) |
| H2B | 0.6532 | 0.1782 | 0.2702 | 0.016* | 0.488 (8) |
| C3A | 0.6609 (2) | 0.3118 (11) | 0.3817 (3) | 0.0133 (10) | 0.488 (8) |
| H3A | 0.6398 | 0.1369 | 0.3847 | 0.016* | 0.488 (8) |
| H3B | 0.6477 | 0.4875 | 0.3976 | 0.016* | 0.488 (8) |
| C4A | 0.7370 (4) | 0.2761 (15) | 0.4512 (6) | 0.0144 (12) | 0.488 (8) |
| H4A | 0.7502 | 0.1078 | 0.4327 | 0.017* | 0.488 (8) |
| H4B | 0.7579 | 0.4560 | 0.4500 | 0.017* | 0.488 (8) |
| C2B | 0.6391 (3) | 0.4728 (14) | 0.2958 (4) | 0.0166 (10) | 0.512 (8) |
| H2C | 0.6284 | 0.6369 | 0.3188 | 0.020* | 0.512 (8) |
| H2D | 0.6685 | 0.5511 | 0.2845 | 0.020* | 0.512 (8) |
| C3B | 0.6752 (2) | 0.2328 (10) | 0.3678 (3) | 0.0178 (10) | 0.512 (8) |
| H3C | 0.6958 | 0.0937 | 0.3523 | 0.021* | 0.512 (8) |
| H3D | 0.6433 | 0.1209 | 0.3689 | 0.021* | 0.512 (8) |
| C4B | 0.7283 (3) | 0.3639 (15) | 0.4615 (5) | 0.0164 (11) | 0.512 (8) |
| H4C | 0.7067 | 0.4920 | 0.4778 | 0.020* | 0.512 (8) |
| H4D | 0.7574 | 0.4906 | 0.4580 | 0.020* | 0.512 (8) |
| C11 | 0.57579 (8) | 0.1819 (3) | 0.55448 (11) | 0.0175 (3) | |
| O1 | 0.57795 (6) | 0.1374 (2) | 0.62108 (8) | 0.0242 (3) | |
| O2 | 0.53729 (5) | 0.3728 (2) | 0.48986 (7) | 0.0205 (3) | |
| H1 | 0.5000 | 0.5000 | 0.5000 | 0.025* | |
| C12 | 0.61862 (9) | 0.0096 (4) | 0.53898 (12) | 0.0233 (4) | |
| Cl1 | 0.68272 (2) | −0.20835 (9) | 0.63586 (3) | 0.02865 (14) | |
| H12A | 0.6394 (8) | 0.142 (4) | 0.5274 (11) | 0.025 (5)* | |
| H12B | 0.5886 (9) | −0.124 (4) | 0.4859 (12) | 0.030 (5)* | |
| Cl2 | 0.5000 | −0.13463 (15) | 0.2500 | 0.03637 (19) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| N1A | 0.016 (2) | 0.021 (2) | 0.0096 (19) | −0.0001 (15) | 0.0101 (16) | 0.0009 (15) |
| N1B | 0.019 (2) | 0.0124 (17) | 0.0133 (19) | 0.0010 (17) | 0.0135 (16) | 0.0012 (13) |
| C2A | 0.0136 (18) | 0.014 (3) | 0.014 (2) | −0.004 (2) | 0.0089 (17) | −0.002 (2) |
| C3A | 0.021 (2) | 0.012 (2) | 0.014 (2) | 0.0007 (16) | 0.0142 (16) | −0.0019 (16) |
| C4A | 0.015 (2) | 0.013 (3) | 0.018 (3) | 0.001 (2) | 0.0116 (18) | −0.001 (2) |
| C2B | 0.0148 (17) | 0.016 (3) | 0.013 (2) | −0.001 (2) | 0.0060 (16) | −0.004 (2) |
| C3B | 0.022 (2) | 0.015 (2) | 0.016 (2) | −0.0024 (16) | 0.0121 (16) | −0.0041 (17) |
| C4B | 0.018 (2) | 0.014 (3) | 0.017 (2) | 0.002 (2) | 0.0111 (18) | 0.001 (2) |
| C11 | 0.0214 (8) | 0.0133 (8) | 0.0195 (8) | −0.0078 (7) | 0.0138 (7) | −0.0049 (7) |
| O1 | 0.0389 (7) | 0.0210 (6) | 0.0254 (6) | 0.0057 (5) | 0.0263 (6) | 0.0066 (5) |
| O2 | 0.0202 (6) | 0.0267 (6) | 0.0131 (6) | 0.0009 (5) | 0.0098 (5) | 0.0022 (5) |
| C12 | 0.0325 (10) | 0.0195 (9) | 0.0283 (10) | −0.0004 (8) | 0.0241 (9) | 0.0016 (8) |
| Cl1 | 0.0277 (2) | 0.0260 (2) | 0.0362 (3) | 0.00387 (18) | 0.0221 (2) | 0.0033 (2) |
| Cl2 | 0.0290 (3) | 0.0488 (4) | 0.0454 (4) | 0.000 | 0.0302 (3) | 0.000 |
Geometric parameters (Å, º) top
| N1A—C2A | 1.495 (6) | C2B—C3B | 1.495 (8) |
| N1A—H1A1 | 0.8900 | C2B—H2C | 0.9700 |
| N1A—H1A2 | 0.8900 | C2B—H2D | 0.9700 |
| N1A—H1A3 | 0.8900 | C3B—C4B | 1.503 (9) |
| N1B—C2B | 1.487 (6) | C3B—H3C | 0.9700 |
| N1B—H1B1 | 0.8900 | C3B—H3D | 0.9700 |
| N1B—H1B2 | 0.8900 | C4B—C4Bi | 1.521 (15) |
| N1B—H1B3 | 0.8900 | C4B—H4C | 0.9700 |
| C2A—C3A | 1.519 (8) | C4B—H4D | 0.9700 |
| C2A—H2A | 0.9700 | C11—O1 | 1.2284 (18) |
| C2A—H2B | 0.9700 | C11—O2 | 1.2848 (18) |
| C3A—C4A | 1.527 (9) | C11—C12 | 1.512 (2) |
| C3A—H3A | 0.9700 | O2—H1 | 1.23 |
| C3A—H3B | 0.9700 | C12—Cl1 | 1.7799 (19) |
| C4A—C4Ai | 1.517 (16) | C12—H12A | 0.905 (17) |
| C4A—H4A | 0.9700 | C12—H12B | 0.982 (18) |
| C4A—H4B | 0.9700 | | |
| | | |
| C2B—N1B—H1B1 | 109.5 | C3B—C2B—H2C | 109.0 |
| C2B—N1B—H1B2 | 109.5 | N1B—C2B—H2D | 109.0 |
| H1B1—N1B—H1B2 | 109.5 | C3B—C2B—H2D | 109.0 |
| C2B—N1B—H1B3 | 109.5 | H2C—C2B—H2D | 107.8 |
| H1B1—N1B—H1B3 | 109.5 | C2B—C3B—C4B | 111.5 (5) |
| H1B2—N1B—H1B3 | 109.5 | C2B—C3B—H3C | 109.3 |
| N1A—C2A—C3A | 111.2 (5) | C4B—C3B—H3C | 109.3 |
| N1A—C2A—H2A | 109.4 | C2B—C3B—H3D | 109.3 |
| C3A—C2A—H2A | 109.4 | C4B—C3B—H3D | 109.3 |
| N1A—C2A—H2B | 109.4 | H3C—C3B—H3D | 108.0 |
| C3A—C2A—H2B | 109.4 | C3B—C4B—C4Bi | 115.5 (7) |
| H2A—C2A—H2B | 108.0 | C3B—C4B—H4C | 108.4 |
| C2A—C3A—C4A | 111.3 (5) | C4Bi—C4B—H4C | 108.4 |
| C2A—C3A—H3A | 109.4 | C3B—C4B—H4D | 108.4 |
| C4A—C3A—H3A | 109.4 | C4Bi—C4B—H4D | 108.4 |
| C2A—C3A—H3B | 109.4 | H4C—C4B—H4D | 107.5 |
| C4A—C3A—H3B | 109.4 | O1—C11—O2 | 125.19 (14) |
| H3A—C3A—H3B | 108.0 | O1—C11—C12 | 122.78 (15) |
| C4Ai—C4A—C3A | 113.2 (7) | O2—C11—C12 | 112.02 (13) |
| C4Ai—C4A—H4A | 108.9 | C11—O2—H1 | 112 |
| C3A—C4A—H4A | 108.9 | C11—C12—Cl1 | 113.44 (12) |
| C4Ai—C4A—H4B | 108.9 | C11—C12—H12A | 109.2 (10) |
| C3A—C4A—H4B | 108.9 | Cl1—C12—H12A | 106.8 (11) |
| H4A—C4A—H4B | 107.7 | C11—C12—H12B | 107.3 (10) |
| N1B—C2B—C3B | 113.0 (5) | Cl1—C12—H12B | 109.5 (10) |
| N1B—C2B—H2C | 109.0 | H12A—C12—H12B | 110.7 (14) |
| | | |
| N1A—C2A—C3A—C4A | 175.6 (4) | C2B—C3B—C4B—C4Bi | −175.5 (6) |
| C2A—C3A—C4A—C4Ai | −176.8 (5) | O1—C11—C12—Cl1 | −11.2 (2) |
| N1B—C2B—C3B—C4B | −166.0 (4) | O2—C11—C12—Cl1 | 169.62 (11) |
| Symmetry code: (i) −x+3/2, −y+1/2, −z+1. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1A—H1A1···Cl2 | 0.89 | 2.12 | 3.003 (6) | 171 |
| N1A—H1A3···Cl2ii | 0.89 | 2.18 | 3.059 (7) | 167 |
| N1A—H1A2···O2iii | 0.89 | 2.21 | 2.988 (7) | 146 |
| N1A—H1A2···O1iv | 0.89 | 2.55 | 3.006 (7) | 113 |
| N1B—H1B3···Cl2 | 0.89 | 2.58 | 3.366 (5) | 147 |
| N1B—H1B1···O1iv | 0.89 | 1.90 | 2.767 (6) | 164 |
| N1B—H1B2···O1v | 0.89 | 1.99 | 2.761 (6) | 144 |
| N1B—H1B2···O2iii | 0.89 | 2.42 | 2.922 (6) | 116 |
| O2—H1···O2vi | 1.23 | 1.23 | 2.452 (1) | 180 |
| Symmetry codes: (ii) x, y+1, z; (iii) −x+1, y, −z+1/2; (iv) x, −y, z−1/2; (v) x, −y+1, z−1/2; (vi) −x+1, −y+1, −z+1. |
Experimental details
| Crystal data |
| Chemical formula | C6H18N22+·C4H5Cl2O4−·Cl− |
| Mr | 341.65 |
| Crystal system, space group | Monoclinic, C2/c |
| Temperature (K) | 100 |
| a, b, c (Å) | 25.702 (5), 4.4419 (4), 18.718 (4) |
| β (°) | 129.14 (3) |
| V (Å3) | 1657.4 (9) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.56 |
| Crystal size (mm) | 0.3 × 0.3 × 0.2 |
| |
| Data collection |
| Diffractometer | Xcalibur, Eos
diffractometer |
| Absorption correction | – |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 2850, 1614, 1301 |
| Rint | 0.027 |
| (sin θ/λ)max (Å−1) | 0.633 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.029, 0.062, 1.05 |
| No. of reflections | 1614 |
| No. of parameters | 135 |
| No. of restraints | 2 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.20, −0.33 |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1A—H1A1···Cl2 | 0.89 | 2.12 | 3.003 (6) | 171.2 |
| N1A—H1A3···Cl2i | 0.89 | 2.18 | 3.059 (7) | 167.1 |
| N1A—H1A2···O2ii | 0.89 | 2.21 | 2.988 (7) | 145.5 |
| N1A—H1A2···O1iii | 0.89 | 2.55 | 3.006 (7) | 112.6 |
| N1B—H1B3···Cl2 | 0.89 | 2.58 | 3.366 (5) | 147.0 |
| N1B—H1B1···O1iii | 0.89 | 1.90 | 2.767 (6) | 163.6 |
| N1B—H1B2···O1iv | 0.89 | 1.99 | 2.761 (6) | 144.3 |
| N1B—H1B2···O2ii | 0.89 | 2.42 | 2.922 (6) | 116.0 |
| O2—H1···O2v | 1.23 | 1.23 | 2.452 (1) | 180 |
| Symmetry codes: (i) x, y+1, z; (ii) −x+1, y, −z+1/2; (iii) x, −y, z−1/2; (iv) x, −y+1, z−1/2; (v) −x+1, −y+1, −z+1. |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](https://journals.iucr.org/c/issues/2009/11/00/fn3035/fn3035fig1thm.gif)
![[Figure 2]](https://journals.iucr.org/c/issues/2009/11/00/fn3035/fn3035fig2thm.gif)
![[Figure 3]](https://journals.iucr.org/c/issues/2009/11/00/fn3035/fn3035fig3thm.gif)




α,ω-Diamino aliphatic alkanes have predictable basic properties because of the two amine groups at the ends of the aliphatic chain. They react with both organic and inorganic acids, resulting in salts composed of a diamine dication and suitable anion(s). The flexible cation tends to adopt a variety of conformations, which are influenced by the crystal packing. This property can be utilized in crystal engineering for synthesis of supramolecular synthons, which control molecular aggregation and lead to particular patterns and new properties (Mahmoudkhani & Langer, 2001\, 2002\), and furthermore for designing new materials possessing, for example, a layered structure (cf. Pospieszna-Markiewicz et al., 2006\).
In the Cambridge Structural Database (CSD; Allen, 2002\; Version 5.30 of October 2008, no multiple entries, no errors), there are 106 crystal structures containing the hexane-1,6-diaminium cation – 38 organic and 68 organometallic. Among the organic structures, there are 27 cases with the dication in a fully extended conformation. There is no particular preference for any of the other possible conformations and none of them is represented by more than three examples. In 18 cases for the organic structures the dications are situated about an inversion center, and in four cases, some other special position. There are examples of structures having a disordered dication (e.g. Freeman, 1984\; Visi et al., 2006\; Yang et al., 2007\; Wang et al., 2006\), anion (Todd & Harrison, 2007\) or even solvent molecule (Liu et al., 2004\; Rather et al., 2003\).
In the course of our studies on the salts of α,ω-diamino aliphatic alkanes with the derivatives of acetic acid, we have determined the crystal structure of the title compound, (I), at 100 K. It can be compared with the crystal structures of hexane-1,6-diaminium dichloride (Binnie & Robertson, 1949\; redetermined by Borkakoti et al., 1978\); with the recently published structure of hexane-1,6-diaminium bis(chloroacetate) (Ortíz et al. 2008\); and with the results of our previous studies on the crystal packing mode of hexane-1,6-diaminium salts with acetic and di- and trichloroacetic acids (Paul & Kubicki, 2009\).
The dication is symmetrical with the mid-point of the central C—C bond residing on the center of inversion at (3/4, 1/4, 1/2) (Fig. 1). The carbon fragment of the dication is disordered over two positions, with the two chains crossing each other at the center of inversion (Fig. 1). The two alternative positions are almost equally populated; the site occupancy factors refined at 0.506 (11) and 0.494 (11). Both disordered dications have the nearly fully extended conformation. Atom Cl on the chloroacetate anion is located almost in the plane of the carboxylate group, with O—C—C—Cl torsion angles of -10.9 (3) and 169.8 (1)°, which is in good agreement with all ten organic crystal structures containing the chloroacetate anion found in the CSD (no multiple entries, no errors, only organic).
The bond distances in the carbon chain of the dication present a similar pattern to those found in hexane-1,6-diaminium dichloride (Binnie & Robertson, 1949\, Borkakoti et al., 1978\), in which there is an alternation of C—C bond lengths. Such a pattern was not observed, however, in the structure of hexane-1,6-diaminium bis(chloroacetate) (Ortíz et al., 2008\). The structure of (I) was determined at 100 K, and the differences in lengths between the neighbouring bonds are still slightly observable; however, they do not meet the standard test for difference (3 s.u.) and are probably the result of the disorder of the aliphatic chain. The average C—C bond length s.u. is 0.004 Å in the report of Borkakoti et al. but averages 0.012 Å in this work. It should also be noted that the s.u. values on the C4—C4' bond across the inversion center are very long at 0.018 Å, suggesting some disorder. No restraints were used in the refinement of the dication.
Carboxylic acid moieties related by the center of inversion at (1/2, 1/2, 1/2) form a hydrogen-bonded dimeric monoanion (cf. Fig. 1), in which one carboxylate group is neutral and one negatively charged. The O—H···O hydrogen bond is short, with an O···O distance of 2.452 (3) Å, with the H atom centered on the inversion center. Refinement resulted in a relatively high isotropic displacement parameter. This suggests that in fact it is disordered between two positions. This is another example of an acid salt, MHX2, of a monocarboxylic acid, HX, belonging to type A (Speakman & Mills, 1961\; Ichikawa, 1972\), with a short hydrogen bond and an H atom situated on an inversion center. The mean bond-length value [2.466 (3) Å] given by Speakman & Mills is in good agreement with our results. The two C—O bond lengths are different [1.287 (2) and 1.224 (2) Å], with the shorter bond not involved in the hydrogen bonding. The difference Fourier map of this region without any H atom clearly shows the significant spread of the residual electron density between the centrosymmetric pair of O atoms.
The three-dimensional network of hydrogen bonds is, along with the electrostatic interactions between the charged species, the main building force in the crystal structure of (I) (Fig. 2). There are three strong bonds between the NH donor groups and oxygen or chloride acceptors (Table 2). The one weaker N—H···O bond can probably be regarded as a secondary interaction, with unfavourable N—H···O angles of ca 119°. The Cl atom from the chloroacetate anion does not participate in hydrogen-bond formation, as was observed by Ortíz et al. (2008\).
Similar interactions were found in the dichloride [N···Cl ranging from 3.145 (3) to 3.191 (2) Å] and in the bis(chloroacetate) [N···O ranging from 2.739 (2) to 2.814 (2) Å]. Likewise, in the series of tri-, di- and acetate salts, three stonger N—H···O and two weaker N—H···Cl/O interactions were observed.
Because of the dication disorder, the hydrogen-bond network of each component should be treated separately. Using graph-set notation (Etter et al., 1990\; Bernstein et al., 1995\), there is an R42(8) ring motif typical for diaminium salts, found only for the 'A' component. However, the other typical motif – R44(26), which is present in tri-, di-, mono- and acetic acid salts – is absent from both forms. Similar to the structure of the bis(chloroacetate), (I) can be described as a molecular ladder, with parallel pair of C43(14) (form 'A') or C44(16) (form 'B') chains acting as the uprights of the ladder, and the cations providing the rungs. Such a ladder can be observed running along the [001] direction (Fig. 2). The ends of the dicationic chains lying in the same layer are linked via two sets of N1A—H1A1···Cl2 and N1A—H1A3···Cl2i (form 'A') or one set of N1B—H1B3···Cl2 (form 'B') hydrogen bonds (symmetry codes as in Table 1) so that the `upper' part of one cation is connected with the `lower' part of the second cation. The different layers are connected via two sets of N1A—H1A2···O1iii and N1A—H1A2···O2ii (form 'A') or three sets of N1B—H1B1···O1iii, N1B—H1B2···O1iv and N1B—H1B2···O2ii (form 'B') hydrogen bonds, where the last interaction of each form is weak and probably secondary. The space-filling representation of the structure (Fig. 3) highlights the alternating layers of dications and anions, similar to those found in the course of studies of the parallel series of hexane-1,6-diaminium salts (Paul & Kubicki, 2009\).