Buy article online - an online subscription or single-article purchase is required to access this article.
The title compound, {(C
6H
14N
2)[Ag(NCS)
3]}
n, is a polymeric silver(I) complex. The Ag
I atom is hexacoordinated by the S atoms of six thiocyanate anions, with each thiocyanate S atom acting in a bridging mode to link the Ag atoms together. The unique Ag
I atom lies at a cell origin and has crystallographically imposed
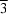
symmetry. The diazonia[2.2.2]octane molecule lies about a site with
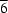
imposed symmetry with the unique N atom on a threefold axis. The S and N atoms of the thiocyanate ligands sit on a mirror plane and a threefold axis, respectively. The crystal structure consists of one-dimensional chains, which are stabilized by N—H

N hydrogen bonds to form a three-dimensional network.
Supporting information
CCDC reference: 251294
AgNO3 (0.170 g, 1 mmol) and 1,4-diazabicyclo[2.2.2]octane (0.5 ml) were dissolved in ammonia solution (10 ml, 30%), and the mixture was stirred for about 30 min at room temperature. The resulting clear colourless solution was allowed to stand in air and, after slow evaporation of the solvent over 15 d, large colourless crystals of (I) were formed at the bottom of the vessel. The crystals were isolated, washed three times with water and dried in a vacuum desiccator using CaCl2 (yield 52.8%). Analysis, found: C 27.32, H 3.48, N 17.62, S 24.31%; calculated for C9H14AgN5S3: C 27.28, H 3.56, N 17.67, S 24.27%.
All H atoms were located in difference Fourier maps and were refined isotropically.
Data collection: SMART (Siemens, 1996); cell refinement: SMART; data reduction: SAINT (Siemens, 1996); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997a); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997a); molecular graphics: SHELXTL (Sheldrick, 1997b); software used to prepare material for publication: SHELXTL.
catena-Poly[1,4-diazonia[2.2.2]octane [silver(I)-tri-µ-thiocyanato-
κ6S:
S]]
top
Crystal data top
| (C6H14N2)[Ag(NCS)3] | Dx = 2.030 Mg m−3 |
| Mr = 396.30 | Mo Kα radiation, λ = 0.71073 Å |
| Hexagonal, P63/m | Cell parameters from 506 reflections |
| a = 10.2148 (14) Å | θ = 3–28° |
| c = 7.1740 (14) Å | µ = 2.03 mm−1 |
| V = 648.26 (18) Å3 | T = 293 K |
| Z = 2 | Needle, colourless |
| F(000) = 396 | 0.30 × 0.10 × 0.08 mm |
Data collection top
Siemens SMART CCD area-detector
diffractometer | 506 independent reflections |
| Radiation source: fine-focus sealed tube | 489 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.160 |
| ϕ and ω scans | θmax = 27.0°, θmin = 2.3° |
Absorption correction: multi-scan
(SADABS; Sheldrick, 1996) | h = −13→8 |
| Tmin = 0.582, Tmax = 0.855 | k = −12→13 |
| 3024 measured reflections | l = −9→8 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.067 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.168 | All H-atom parameters refined |
| S = 1.06 | w = 1/[σ2(Fo2) + (0.0632P)2 + 3.05P]
where P = (Fo2 + 2Fc2)/3 |
| 506 reflections | (Δ/σ)max < 0.001 |
| 43 parameters | Δρmax = 0.81 e Å−3 |
| 1 restraint | Δρmin = −0.69 e Å−3 |
Crystal data top
| (C6H14N2)[Ag(NCS)3] | Z = 2 |
| Mr = 396.30 | Mo Kα radiation |
| Hexagonal, P63/m | µ = 2.03 mm−1 |
| a = 10.2148 (14) Å | T = 293 K |
| c = 7.1740 (14) Å | 0.30 × 0.10 × 0.08 mm |
| V = 648.26 (18) Å3 | |
Data collection top
Siemens SMART CCD area-detector
diffractometer | 506 independent reflections |
Absorption correction: multi-scan
(SADABS; Sheldrick, 1996) | 489 reflections with I > 2σ(I) |
| Tmin = 0.582, Tmax = 0.855 | Rint = 0.160 |
| 3024 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.067 | 1 restraint |
| wR(F2) = 0.168 | All H-atom parameters refined |
| S = 1.06 | Δρmax = 0.81 e Å−3 |
| 506 reflections | Δρmin = −0.69 e Å−3 |
| 43 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Ag1 | 1.0000 | 1.0000 | 0.0000 | 0.0510 (5) | |
| S1 | 0.7928 (2) | 0.7865 (2) | 0.2500 | 0.0352 (6) | |
| N2 | 0.3333 | 0.6667 | 0.0774 (9) | 0.0213 (14) | |
| C2 | 0.1800 (5) | 0.6260 (6) | 0.1463 (7) | 0.0256 (11) | |
| N1 | 0.8424 (8) | 0.5408 (8) | 0.2500 | 0.0379 (16) | |
| C1 | 0.8227 (8) | 0.6425 (8) | 0.2500 | 0.0268 (15) | |
| H1 | 0.115 (5) | 0.538 (6) | 0.104 (7) | 0.009 (11)* | |
| H2 | 0.163 (6) | 0.690 (7) | 0.092 (8) | 0.022 (13)* | |
| H3 | 0.3333 | 0.6667 | −0.050 (3) | 0.03 (3)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Ag1 | 0.0514 (6) | 0.0514 (6) | 0.0503 (8) | 0.0257 (3) | 0.000 | 0.000 |
| S1 | 0.0249 (10) | 0.0292 (10) | 0.0530 (12) | 0.0145 (8) | 0.000 | 0.000 |
| N2 | 0.019 (2) | 0.019 (2) | 0.025 (3) | 0.0097 (10) | 0.000 | 0.000 |
| C2 | 0.016 (2) | 0.027 (2) | 0.033 (3) | 0.010 (2) | −0.0035 (19) | 0.001 (2) |
| N1 | 0.040 (4) | 0.031 (4) | 0.040 (3) | 0.015 (3) | 0.000 | 0.000 |
| C1 | 0.022 (3) | 0.028 (4) | 0.027 (3) | 0.011 (3) | 0.000 | 0.000 |
Geometric parameters (Å, º) top
| Ag1—S1i | 2.7993 (15) | N2—C2 | 1.490 (5) |
| Ag1—S1ii | 2.7993 (15) | N2—C2vii | 1.490 (5) |
| Ag1—S1iii | 2.7993 (15) | N2—C2viii | 1.490 (5) |
| Ag1—S1iv | 2.7993 (15) | N2—H3 | 0.91 (2) |
| Ag1—S1v | 2.7993 (15) | C2—C2ix | 1.489 (10) |
| Ag1—S1 | 2.7993 (15) | C2—H1 | 0.86 (5) |
| S1—C1 | 1.645 (8) | C2—H2 | 0.86 (6) |
| S1—Ag1vi | 2.7993 (15) | N1—C1 | 1.152 (10) |
| | | |
| S1i—Ag1—S1ii | 96.65 (4) | C1—S1—Ag1 | 106.89 (19) |
| S1i—Ag1—S1iii | 83.35 (4) | Ag1vi—S1—Ag1 | 79.69 (5) |
| S1ii—Ag1—S1iii | 180.0 | C2—N2—C2vii | 109.6 (3) |
| S1i—Ag1—S1iv | 96.65 (4) | C2—N2—C2viii | 109.6 (3) |
| S1ii—Ag1—S1iv | 83.35 (4) | C2vii—N2—C2viii | 109.6 (3) |
| S1iii—Ag1—S1iv | 96.65 (4) | C2—N2—H3 | 109.4 (3) |
| S1i—Ag1—S1v | 180.0 | C2vii—N2—H3 | 109.4 (3) |
| S1ii—Ag1—S1v | 83.35 (4) | C2viii—N2—H3 | 109.4 (3) |
| S1iii—Ag1—S1v | 96.65 (4) | C2ix—C2—N2 | 109.4 (3) |
| S1iv—Ag1—S1v | 83.35 (4) | C2ix—C2—H1 | 111 (3) |
| S1i—Ag1—S1 | 83.35 (4) | N2—C2—H1 | 109 (3) |
| S1ii—Ag1—S1 | 96.65 (4) | C2ix—C2—H2 | 117 (4) |
| S1iii—Ag1—S1 | 83.35 (4) | N2—C2—H2 | 103 (4) |
| S1iv—Ag1—S1 | 180.0 | H1—C2—H2 | 107 (5) |
| S1v—Ag1—S1 | 96.65 (4) | N1—C1—S1 | 179.5 (7) |
| C1—S1—Ag1vi | 106.89 (19) | | |
| | | |
| S1i—Ag1—S1—C1 | −62.66 (19) | S1iii—Ag1—S1—Ag1vi | −42.023 (15) |
| S1ii—Ag1—S1—C1 | 33.30 (18) | S1iv—Ag1—S1—Ag1vi | −99.40 (12) |
| S1iii—Ag1—S1—C1 | −146.70 (18) | S1v—Ag1—S1—Ag1vi | −137.977 (15) |
| S1iv—Ag1—S1—C1 | 156 (46) | C2vii—N2—C2—C2ix | −60.1 (4) |
| S1v—Ag1—S1—C1 | 117.34 (19) | C2viii—N2—C2—C2ix | 60.1 (4) |
| S1i—Ag1—S1—Ag1vi | 42.023 (15) | Ag1vi—S1—C1—N1 | 137.96 (6) |
| S1ii—Ag1—S1—Ag1vi | 137.977 (15) | Ag1—S1—C1—N1 | −137.96 (6) |
| Symmetry codes: (i) −y+2, x−y+1, z; (ii) x−y+1, x, −z; (iii) −x+y+1, −x+2, z; (iv) −x+2, −y+2, −z; (v) y, −x+y+1, −z; (vi) −x+2, −y+2, z+1/2; (vii) −x+y, −x+1, z; (viii) −y+1, x−y+1, z; (ix) x, y, −z+1/2. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H3···N1x | 0.91 (2) | 2.44 (1) | 3.070 (7) | 126 (1) |
| Symmetry code: (x) −x+1, −y+1, −z. |
Experimental details
| Crystal data |
| Chemical formula | (C6H14N2)[Ag(NCS)3] |
| Mr | 396.30 |
| Crystal system, space group | Hexagonal, P63/m |
| Temperature (K) | 293 |
| a, c (Å) | 10.2148 (14), 7.1740 (14) |
| V (Å3) | 648.26 (18) |
| Z | 2 |
| Radiation type | Mo Kα |
| µ (mm−1) | 2.03 |
| Crystal size (mm) | 0.30 × 0.10 × 0.08 |
| |
| Data collection |
| Diffractometer | Siemens SMART CCD area-detector
diffractometer |
| Absorption correction | Multi-scan
(SADABS; Sheldrick, 1996) |
| Tmin, Tmax | 0.582, 0.855 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 3024, 506, 489 |
| Rint | 0.160 |
| (sin θ/λ)max (Å−1) | 0.638 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.067, 0.168, 1.06 |
| No. of reflections | 506 |
| No. of parameters | 43 |
| No. of restraints | 1 |
| H-atom treatment | All H-atom parameters refined |
| Δρmax, Δρmin (e Å−3) | 0.81, −0.69 |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H3···N1i | 0.91 (2) | 2.443 (14) | 3.070 (7) | 125.9 (4) |
| Symmetry code: (i) −x+1, −y+1, −z. |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu metal-organic compounds
metal-organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2004/09/00/av1193/av1193fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2004/09/00/av1193/av1193fig2thm.gif)

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry



Because silver(I) is good candidate as a soft acid favouring coordination to soft bases, such as ligands containing S and N atoms, we and others have exploited the coordination flexibility of silver(I) in the construction of a large number of coordination polymers, which exhibit interesting structural diversity (Yang et al., 2000; Zhu Zhang et al., 2003; Zhu et al., 2004). Thiocyanate is a potential bridging ligand. Thus, many complexes containing AgSCN units have been reported (Cotton & Wilkinson, 1988; Krautscheid et al., 1998; Ren et al., 2001). In this paper we report the title novel silver(I) thiocyanate salt, (I). \sch
Compound (I) is a polymeric silver(I) complex. The AgI atoms sit on a sixfold axis, and their site occupancy factors are 1/6 or 0.16667. The other 5/6 of each atom is generated by the symmetry elements that pass through those special positons. The S and N atoms of the thiocyanate ligands sit on the mirror plane and threefold axis, respectively, with site occupancy factors of 1/2 and 1/3, respectively.
The Ag atom is pseudo-octahedrally six-coordinated by three pairs of bridging S atoms which link the Ag atoms together, forming a one-dimensional polymeric chain (Fig. 1). The smallest repeat unit consists of a silver-thiocyanate-1,4-diazonia[2.2.2]octane adduct in 1:3:1 stoichiometry, in which 1,4-diazonia[2.2.2]octane acts as a cation, not as a coordination ligand to the Ag atom. The six Ag—S bonds are the same [2.7993 (15) Å] and are normal by comparison with those in a similar complex (Zhu Liu & Meng, 2003). The bond angles are in the range 83.35 (4)–96.65 (4)° at the Ag atom, so each Ag atom lies almost in the centre of a slightly distorted octahedron. The thiocyanate group is almost linear [S—C 1.645 (8) and C—N 1.490 (5) Å, and S—C—N 179.5 (7)°]. The Ag—S—Ag angle is 79.69 (5)°. The remaining C—S—Ag angles about the S atom are both 106.89 (19)°, so that the geometry at the S atom is a slightly distorted pyramid.
In the crystal of (I), the 1,4-diazonia[2.2.2]octane cations are located between the chains. The bridging S atoms link the Ag atoms into a linear chain along the c axis. Adjacent chains interact with the cations via three equivalent hydrogen bonds [N2—H3 0.91 (2), H3···A 2.443 (14) and N2···A 3.070 (7) Å, and N2—H3···A 125.9 (4)°, where A is N1i, N1ii and N1iii; symmetry codes: (i) 1 − x, 1 − y, −z; (ii) x-y, x, −z; (iii) y, 1 − x + y, −z], forming a three-dimensional structure (Fig.2)