

 journal menu
journal menu organic compounds
organic compounds






In the hydrogen-bond patterns of phenyl bis(2-chlorobenzylamido)phosphinate, C20H19Cl2N2O2P, (I), and N,N'-bis(2-chlorobenzyl)-N''-(2,2,2-trifluoroacetyl)phosphoric triamide, C16H15Cl2F3N3O2P, (II), the O atoms of the related phosphoryl groups act as double H-atom acceptors, so that the P=O (H-N)2 hydrogen bond in (I) and the P=O(H-Namide)2 and C=OH-NC(O)NHP(O) hydrogen bonds in (II) are responsible for the aggregation of the molecules in the crystal packing. The presence of a double H-atom acceptor centre is a result of the involvement of a greater number of H-atom donor sites with a smaller number of H-atom acceptor sites in the hydrogen-bonding interactions. This article also reviews structures having a P(O)NH group, with the aim of finding similar three-centre hydrogen bonds in the packing of phosphoramidate compounds. This analysis shows that the factors affecting the preference of the above-mentioned O atom to act as a double H-atom acceptor are: (i) a higher number of H-atom donor sites relative to H-atom acceptor centres in molecules with P(=O)(NH)3, (N)P(=O)(NH)2, C(=O)NHP(=O)(NH)2 and (NH)2P(=O)OP(=O)(NH)2 groups, and (ii) the remarkable H-atom acceptability of this atom relative to the other acceptor centre(s) in molecules containing an OP(=O)(NH)2 group, with the explanation that the N atom bound to the P atom in almost all of the structures found does not take part in hydrogen bonding as an acceptor. Moreover, the differences in the H-atom acceptability of the phosphoryl O atom relative to the O atom of the alkoxy or phenoxy groups in amidophosphoric acid esters may be illustrated by considering the molecular packing of compounds having (O)2P(=O)(NH) and (O)P(=O)(NH)(N)groups, in which the unique N-H unit in the above-mentioned molecules almost always selects the phosphoryl O atom as a partner in forming hydrogen-bond interactions. The P atoms in (I) and (II) are in tetrahedral coordination environments, and the phosphoryl and carbonyl groups in (II) are anti with respect to each other (the P and C groups are separated by one N atom). In the crystal structures of (I) and (II), adjacent molecules are linked via the above-mentioned hydrogen bonds into a linear arrangement parallel to [100] in both cases, in (I) by forming R22(8) rings and in (II) through a combination of R22(10) and R21(6) rings.
(H-N)2 hydrogen bond in (I) and the P=O(H-Namide)2 and C=OH-NC(O)NHP(O) hydrogen bonds in (II) are responsible for the aggregation of the molecules in the crystal packing. The presence of a double H-atom acceptor centre is a result of the involvement of a greater number of H-atom donor sites with a smaller number of H-atom acceptor sites in the hydrogen-bonding interactions. This article also reviews structures having a P(O)NH group, with the aim of finding similar three-centre hydrogen bonds in the packing of phosphoramidate compounds. This analysis shows that the factors affecting the preference of the above-mentioned O atom to act as a double H-atom acceptor are: (i) a higher number of H-atom donor sites relative to H-atom acceptor centres in molecules with P(=O)(NH)3, (N)P(=O)(NH)2, C(=O)NHP(=O)(NH)2 and (NH)2P(=O)OP(=O)(NH)2 groups, and (ii) the remarkable H-atom acceptability of this atom relative to the other acceptor centre(s) in molecules containing an OP(=O)(NH)2 group, with the explanation that the N atom bound to the P atom in almost all of the structures found does not take part in hydrogen bonding as an acceptor. Moreover, the differences in the H-atom acceptability of the phosphoryl O atom relative to the O atom of the alkoxy or phenoxy groups in amidophosphoric acid esters may be illustrated by considering the molecular packing of compounds having (O)2P(=O)(NH) and (O)P(=O)(NH)(N)groups, in which the unique N-H unit in the above-mentioned molecules almost always selects the phosphoryl O atom as a partner in forming hydrogen-bond interactions. The P atoms in (I) and (II) are in tetrahedral coordination environments, and the phosphoryl and carbonyl groups in (II) are anti with respect to each other (the P and C groups are separated by one N atom). In the crystal structures of (I) and (II), adjacent molecules are linked via the above-mentioned hydrogen bonds into a linear arrangement parallel to [100] in both cases, in (I) by forming R22(8) rings and in (II) through a combination of R22(10) and R21(6) rings.
(H-N)2 hydrogen bond in (I) and the P=O(H-Namide)2 and C=OH-NC(O)NHP(O) hydrogen bonds in (II) are responsible for the aggregation of the molecules in the crystal packing. The presence of a double H-atom acceptor centre is a result of the involvement of a greater number of H-atom donor sites with a smaller number of H-atom acceptor sites in the hydrogen-bonding interactions. This article also reviews structures having a P(O)NH group, with the aim of finding similar three-centre hydrogen bonds in the packing of phosphoramidate compounds. This analysis shows that the factors affecting the preference of the above-mentioned O atom to act as a double H-atom acceptor are: (i) a higher number of H-atom donor sites relative to H-atom acceptor centres in molecules with P(=O)(NH)3, (N)P(=O)(NH)2, C(=O)NHP(=O)(NH)2 and (NH)2P(=O)OP(=O)(NH)2 groups, and (ii) the remarkable H-atom acceptability of this atom relative to the other acceptor centre(s) in molecules containing an OP(=O)(NH)2 group, with the explanation that the N atom bound to the P atom in almost all of the structures found does not take part in hydrogen bonding as an acceptor. Moreover, the differences in the H-atom acceptability of the phosphoryl O atom relative to the O atom of the alkoxy or phenoxy groups in amidophosphoric acid esters may be illustrated by considering the molecular packing of compounds having (O)2P(=O)(NH) and (O)P(=O)(NH)(N)groups, in which the unique N-H unit in the above-mentioned molecules almost always selects the phosphoryl O atom as a partner in forming hydrogen-bond interactions. The P atoms in (I) and (II) are in tetrahedral coordination environments, and the phosphoryl and carbonyl groups in (II) are anti with respect to each other (the P and C groups are separated by one N atom). In the crystal structures of (I) and (II), adjacent molecules are linked via the above-mentioned hydrogen bonds into a linear arrangement parallel to [100] in both cases, in (I) by forming R22(8) rings and in (II) through a combination of R22(10) and R21(6) rings.organic compounds
In N,N′-di-tert-butyl-N′′,N′′-dimethylphosphoric triamide, C10H26N3OP, (I), and N,N′,N′′,N′′′-tetra-tert-butoxybis(phosphonic diamide), C16H40N4O3P2, (II), the extended structures are mediated by P(O)(H—N)2 interactions. The asymmetric unit of (I) consists of six independent molecules which aggregate through P(O)(H—N)2 hydrogen bonds, giving R21(6) loops and forming two independent chains parallel to the a axis. Of the 12 independent tert-butyl groups, five are disordered over two different positions with occupancies ranging from 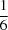 to
to 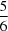 . In the structure of (II), the asymmetric unit contains one molecule. P(O)(H—N)2 hydrogen bonds give S(6) and R22(8) rings, and the molecules form extended chains parallel to the c axis. The structures of (I) and (II), along with similar structures having (N)P(O)(NH)2 and (NH)2P(O)(O)P(O)(NH)2 skeletons extracted from the Cambridge Structural Database, are used to compare hydrogen-bond patterns in these families of phosphoramidates. The strengths of P(O)[H—N]x (x = 1, 2 or 3) hydrogen bonds are also analysed, using these compounds and previously reported structures with (N)2P(O)(NH) and P(O)(NH)3 fragments.
. In the structure of (II), the asymmetric unit contains one molecule. P(O)(H—N)2 hydrogen bonds give S(6) and R22(8) rings, and the molecules form extended chains parallel to the c axis. The structures of (I) and (II), along with similar structures having (N)P(O)(NH)2 and (NH)2P(O)(O)P(O)(NH)2 skeletons extracted from the Cambridge Structural Database, are used to compare hydrogen-bond patterns in these families of phosphoramidates. The strengths of P(O)[H—N]x (x = 1, 2 or 3) hydrogen bonds are also analysed, using these compounds and previously reported structures with (N)2P(O)(NH) and P(O)(NH)3 fragments.
(H—N)2 interactions. The asymmetric unit of (I) consists of six independent molecules which aggregate through P(O)(H—N)2 hydrogen bonds, giving R21(6) loops and forming two independent chains parallel to the a axis. Of the 12 independent tert-butyl groups, five are disordered over two different positions with occupancies ranging from (H—N)2 hydrogen bonds give S(6) and R22(8) rings, and the molecules form extended chains parallel to the c axis. The structures of (I) and (II), along with similar structures having (N)P(O)(NH)2 and (NH)2P(O)(O)P(O)(NH)2 skeletons extracted from the Cambridge Structural Database, are used to compare hydrogen-bond patterns in these families of phosphoramidates. The strengths of P(O)[H—N]x (x = 1, 2 or 3) hydrogen bonds are also analysed, using these compounds and previously reported structures with (N)2P(O)(NH) and P(O)(NH)3 fragments.organic compounds
In the phosphoric triamides N,N,N′,N′-tetrabenzyl-N′′-(2-chloro-2,2-difluoroacetyl)phosphoric triamide, C30H29ClF2N3O2P, (I), N,N,N′,N′-tetrabenzyl-N′′-(3-fluorobenzoyl)phosphoric triamide, C35H33FN3O2P, (II), and N,N,N′,N′-tetrabenzyl-N′′-(3,5-difluorobenzoyl)phosphoric triamide, C35H32F2N3O2P, (III), the tertiary N atoms of the dibenzylamido groups have sp2 character with minimal deviation from planarity. The sums of the three bond angles about the N atoms in (I)–(III) deviate by less than 8° from the planar value of 360°. The geometries of the tertiary N atoms in all phosphoric triamides with C(O)NHP(O)[N]2 skeletons deposited in the Cambridge Structural Database [CSD; Allen (2002). Acta Cryst. B58, 380–388] have been examined and the bond-angle sums at the two tertiary N atoms (SUM1 and SUM2) and the parameter ΔSUM (= SUM1 − SUM2) considered. It was found that in compounds with a considerable ΔSUM value, the more pyramidal N atoms are usually oriented so that the corresponding lone electron pair is anti with respect to the P=O group. In (I), (II) and (III), the phosphoryl and carbonyl groups, separated by an N atom, are anti with respect to each other. In the C(O)NHP(O) fragment of (I)–(III), the P—N bond is longer and the O—P—N angle is contracted compared with the other two P—N bonds and the O—P—N angles in the molecules. These effects are also seen in analogous compounds deposited in the CSD. Compounds with [C(O)NH]P(O)[N]X (X ≠ N), such as compounds with a [C(O)NH]P(O)[N][O] skeleton, have not been considered here. Also, compounds with a [C(O)NH]2P(O)[N] fragment have not been reported to date. In the crystal structures of all three title compounds, adjacent molecules are linked via pairs of P=OH—N hydrogen bonds, forming dimers with Ci symmetry.
H—N hydrogen bonds, forming dimers with Ci symmetry.


