

 journal menu
journal menu metal-organic compounds
metal-organic compounds






The title compound, [Zn(C19H12N5)2], crystallizes in the tetragonal space group P43212, with the monomer residing on a twofold axis. The imidazole N-bound H atoms are disordered over the two positions, with refined occupancies of 0.59 (3) and 0.41 (3). The strong similarities to, and slight differences from, a reported P42212 polymorph which has a 50% smaller unit-cell volume [Harvey, Baggio, Muñoz & Baggio (2003). Acta Cryst. C59, m283–m285], to which the present structure bears a group–subgroup relationship, are discussed.
metal-organic compounds
The title complex, [Na(C8H9O5S)]n, is polymeric and consists of broad layers parallel to (100) made up of an inner hydrophilic core of Na+ cations and polar SO3C(OH)– groups, padded on both sides by two hydrophobic layers of pendant methoxyphenyl groups. The Na+ cations in the inner core are six-coordinated into highly distorted NaO6 octahedra by four symmetry-related (hydroxy)(4-methoxyphenyl)methanesulfonate anions, leading to a tightly woven two-dimensional structure. While there are some hydrogen bonds providing interplanar cohesion, interactions between adjacent layers are weak hydrophobic ones. The present compound appears to be the first reported structure containing the (hydroxy)(4-methoxyphenyl)methanesulfonate ligand.
organic compounds
The title compound, C17H10F5N5O2, is described and compared with its 4-nitrophenyl isomer [Bustos, Sánchez, Schott, Alvarez-Thon & Fuentealba (2007). Acta Cryst. E63, o1138–o1139]. The title molecule presents its nitro group split into two rotationally disordered components, which in conjunction with the rotation of the `unclamped' rings constitute the main molecular differences. Packing is directed by a head-to-tail type `I' C—F F—C interaction, generating double-chain strips running along [100]. These substructures are interlinked by a variety of weak FF, OF, Fπ and Oπ interactions.
F—C interaction, generating double-chain strips running along [100]. These substructures are interlinked by a variety of weak FF, OF, Fπ and Oπ interactions.
F—C interaction, generating double-chain strips running along [100]. These substructures are interlinked by a variety of weak FF, OF, Fπ and Oπ interactions.organic compounds
The asymmetric unit of the title salt [systematic name: bis(4-(2,3-dichlorophenyl)-1-{4-[(2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)oxy]butyl}piperazin-1-ium) oxalate-oxalic acid (1/1)], 2C23H28Cl2N3O2+·C2O42-·C2H2O4, consists of one protonated aripiprazole unit (HArip+), half an oxalate dianion and half an oxalic acid molecule, the latter two lying on inversion centres. The conformation of the HArip+ cation differs from that in other reported salts and resembles more the conformation of neutral Arip units in reported polymorphs and solvates. The intermolecular interaction linking HArip+ cations is also similar to those in reported Arip compounds crystallizing in the space group P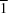 , with head-to-head N-HO hydrogen bonds generating centrosymmetric dimers, which are further organized into planar ribbons parallel to (01
, with head-to-head N-HO hydrogen bonds generating centrosymmetric dimers, which are further organized into planar ribbons parallel to (01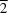 ). The oxalate anions and oxalic acid molecules form hydrogen-bonded chains running along [010], which `pierce' the planar ribbons, interacting with them through a number of stronger N-HO and weaker C-HO hydrogen bonds, forming a three-dimensional network.
). The oxalate anions and oxalic acid molecules form hydrogen-bonded chains running along [010], which `pierce' the planar ribbons, interacting with them through a number of stronger N-HO and weaker C-HO hydrogen bonds, forming a three-dimensional network.
O hydrogen bonds generating centrosymmetric dimers, which are further organized into planar ribbons parallel to (01O and weaker C-HO hydrogen bonds, forming a three-dimensional network.organic compounds
Crystal structures are presented for two members of the homologous series of 1,2-dibromo-4,5-dialkoxybenzenes, viz. those with decyloxy and hexadecyloxy substituents, namely 1,2-dibromo-4,5-bis(decyloxy)benzene, C26H44Br2O2, (II), and 1,2-dibromo-4,5-bis(hexadecyloxy)benzene, C38H68Br2O2, (III). The relative influences which halogen bonding, ![[pi]](/logos/entities/pi_rmgif.gif) - stacking and van der Waals interactions have on these structures are analysed and the results compared with those already found for the lightest homologue, 1,2-dibromo-4,5-dimethoxybenzene, (I) [Cukiernik, Zelcer, Garland & Baggio (2008). Acta Cryst. C64, o604-o608]. The results confirm that the prevalent interactions stabilizing the structures of (II) and (III) are van der Waals contacts between the aliphatic chains. In the case of (II), weak halogen C-Br(Br-C)' interactions are also present and contribute to the stability of the structure. In the case of (III), van der Waals interactions between the aliphatic chains are almost exclusive, weaker C-Br interactions being the only additional interactions detected. The results are in line with commonly accepted models concerning trends in crystal stability along a homologous series (as measured by their melting points), but the earlier report for n = 1, and the present report for n = 10 and 16, are among the few providing single-crystal information validating the hypothesis.
- stacking and van der Waals interactions have on these structures are analysed and the results compared with those already found for the lightest homologue, 1,2-dibromo-4,5-dimethoxybenzene, (I) [Cukiernik, Zelcer, Garland & Baggio (2008). Acta Cryst. C64, o604-o608]. The results confirm that the prevalent interactions stabilizing the structures of (II) and (III) are van der Waals contacts between the aliphatic chains. In the case of (II), weak halogen C-Br(Br-C)' interactions are also present and contribute to the stability of the structure. In the case of (III), van der Waals interactions between the aliphatic chains are almost exclusive, weaker C-Br interactions being the only additional interactions detected. The results are in line with commonly accepted models concerning trends in crystal stability along a homologous series (as measured by their melting points), but the earlier report for n = 1, and the present report for n = 10 and 16, are among the few providing single-crystal information validating the hypothesis.
- stacking and van der Waals interactions have on these structures are analysed and the results compared with those already found for the lightest homologue, 1,2-dibromo-4,5-dimethoxybenzene, (I) [Cukiernik, Zelcer, Garland & Baggio (2008). Acta Cryst. C64, o604-o608]. The results confirm that the prevalent interactions stabilizing the structures of (II) and (III) are van der Waals contacts between the aliphatic chains. In the case of (II), weak halogen C-Br(Br-C)' interactions are also present and contribute to the stability of the structure. In the case of (III), van der Waals interactions between the aliphatic chains are almost exclusive, weaker C-Br interactions being the only additional interactions detected. The results are in line with commonly accepted models concerning trends in crystal stability along a homologous series (as measured by their melting points), but the earlier report for n = 1, and the present report for n = 10 and 16, are among the few providing single-crystal information validating the hypothesis.metal-organic compounds
Poly[[tetraaquadi-μ4-citrato-tetrakis(2,6-diaminopurine)tetracobalt(II)] 6.35-hydrate], {[Co4(C6H4O7)2(C5H6N6)4(H2O)4]·6.35H2O}n, presents three different types of CoII cations in the asymmetric unit, two of them lying on symmetry elements (one on an inversion centre and the other on a twofold axis). The main fragment is further composed of one fully deprotonated citrate (cit) tetraanion, two 2,6-diaminopurine (dap) molecules and two aqua ligands. The structure is completed by a mixture of fully occupied and disordered solvent water molecules. The two independent dap ligands are neutral and the cit tetraanion provides for charge balance, compensating the 4+ cationic charge. There are two well defined coordination geometries in the structure. The simplest is mononuclear, with the CoII cation arranged in a regular centrosymmetric octahedral array, coordinated by two aqua ligands, two dap ligands and two O atoms from the β-carboxylate groups of the bridging cit tetraanions. The second, more complex, group is trinuclear, bisected by a twofold axis, with the metal centres coordinated by two cit tetraanions through their α- and β-carboxylate and α-hydroxy groups, and by two dap ligands bridging through one of their pyridine and one of their imidazole N atoms. The resulting coordination geometry around each metal centre is distorted octahedral. Both groups are linked alternately to each other, defining parallel chains along [201], laterally interleaved and well connected via hydrogen bonding to form a strongly coupled three-dimensional network. The compound presents a novel μ4-κ5O:O,O′:O′,O′′,O′′′:O′′′′ mode of coordination of the cit tetraanion.
metal-organic compounds
The title ionic compound, [Ni(C12H12N2)(H2O)4]SO4·H2O, is composed of an NiII cation coordinated by a chelating 4,4'-dimethyl-2,2'-bipyridine ligand via its two N atoms [mean Ni-N = 2.056 (2) Å] and by four aqua ligands [mean Ni-O = 2.073 (9) Å], the net charge being balanced by an external sulfate anion. The whole structure is stabilized by a solvent water molecule. Even though the individual constituents are rather featureless, they generate an extremely complex supramolecular structure consisting of a central hydrogen-bonded two-dimensional hydrophilic nucleus made up of complex cations, sulfate anions and coordinated and solvent water molecules, with pendant hydrophobic 4,4'-dimethyl-2,2'-bipyridine ligands which interact laterally with their neighbours via - interactions. The structure is compared with closely related analogues in the literature.
- interactions. The structure is compared with closely related analogues in the literature.metal-organic compounds
Tricarbonyl[9-(triphenylphosphonio)fluorenylidene]ruthenium, [Ru(C31H21P)(CO)3], (I), is mononuclear, consisting of a single Ru centre, to which three carbonyl units and a chelating μ3-9-(triphenylphosphonio)fluorenide ylide bind to generate a distorted octahedral RuC6 core. Nonacarbonyl-μ3-fluorenylidene-μ2-hydrido-triangulo-triosmium(III), [Os3H(C13H7)(CO)9], (II), is trinuclear and presents a triangular triosmium core, nine carbonyl ligands and one fluorenylidene ligand. Two of the OsIII centres present a highly distorted hexacoordinated Os(Os2C4) core and are in turn bridged by a hydride ligand. The remaining OsIII cation is octacoordinated, with an Os(Os2C6) nucleus. The crystal structures of both compounds are the result of nondirectional forces, much resembling the packing of weakly interacting quasi-spherical units, viz. the molecules themselves in (I) and centrosymmetric π–π-bonded dimers in (II).
metal-organic compounds
Two copper complex solvatomorphs, namely (3,10-C-meso-3,5,7,7,10,12,14,14-octamethyl-1,4,8,11-tetraazacyclotetradecane)bis(perchlorato-![[kappa]](/logos/entities/kappa_rmgif.gif) O)copper(II) 1.2-hydrate, [Cu(ClO4)2(C18H40N4)]·1.2H2O, (I), and (3,10-C-meso-3,5,7,7,10,12,14,14-octamethyl-1,4,8,11-tetraazacyclotetradecane)bis(perchlorato-O)copper(II), [Cu(ClO4)2(C18H40N4)], (II), are described and compared with each other and with a third, already reported, anhydrous diastereomer, denoted (III). Both compounds present very similar centrosymmetic coordination environments, with the CuII cation lying on an inversion centre in a distorted 4+2 octahedral environment, defined by the macrocyclic N4 group in the equatorial sites and two perchlorate groups in trans-axial positions [one of the perchlorate ligands in (I) is partially disordered]. The most significant difference in molecular shape is seen in the orientation of the perchlorate anions, and the influence of this on the intramolecular hydrogen bonding is discussed. The (partially) hydrated state of (I) favours the formation of chains along [011], while the anhydrous character of (II) and (III) promotes loosely bound structures with low packing indices.
O)copper(II) 1.2-hydrate, [Cu(ClO4)2(C18H40N4)]·1.2H2O, (I), and (3,10-C-meso-3,5,7,7,10,12,14,14-octamethyl-1,4,8,11-tetraazacyclotetradecane)bis(perchlorato-O)copper(II), [Cu(ClO4)2(C18H40N4)], (II), are described and compared with each other and with a third, already reported, anhydrous diastereomer, denoted (III). Both compounds present very similar centrosymmetic coordination environments, with the CuII cation lying on an inversion centre in a distorted 4+2 octahedral environment, defined by the macrocyclic N4 group in the equatorial sites and two perchlorate groups in trans-axial positions [one of the perchlorate ligands in (I) is partially disordered]. The most significant difference in molecular shape is seen in the orientation of the perchlorate anions, and the influence of this on the intramolecular hydrogen bonding is discussed. The (partially) hydrated state of (I) favours the formation of chains along [011], while the anhydrous character of (II) and (III) promotes loosely bound structures with low packing indices.
O)copper(II) 1.2-hydrate, [Cu(ClO4)2(C18H40N4)]·1.2H2O, (I), and (3,10-C-meso-3,5,7,7,10,12,14,14-octamethyl-1,4,8,11-tetraazacyclotetradecane)bis(perchlorato-O)copper(II), [Cu(ClO4)2(C18H40N4)], (II), are described and compared with each other and with a third, already reported, anhydrous diastereomer, denoted (III). Both compounds present very similar centrosymmetic coordination environments, with the CuII cation lying on an inversion centre in a distorted 4+2 octahedral environment, defined by the macrocyclic N4 group in the equatorial sites and two perchlorate groups in trans-axial positions [one of the perchlorate ligands in (I) is partially disordered]. The most significant difference in molecular shape is seen in the orientation of the perchlorate anions, and the influence of this on the intramolecular hydrogen bonding is discussed. The (partially) hydrated state of (I) favours the formation of chains along [011], while the anhydrous character of (II) and (III) promotes loosely bound structures with low packing indices.metal-organic compounds
The isomorphous title compounds, [Tr(S4O6)(C12H12N2)2]·2C3H7NO (Tr = CdII and ZnII), consist of metal centres to which one tetrathionate and two 4,4′-dimethyl-2,2′-bipyridine chelating ligands bind. The structures are completed by two symmetry-related dimethylformamide solvent molecules. Each metal-centred complex is bisected by a twofold axis running through the metal centre and halving the chelating tetrathionate dianion through the central S—S bond. The ancillary symmetry-related 4,4′-dimethyl-2,2′-bipyridine ligands act as chelates. This results in a distorted six-coordinate geometry, with similar Tr—O/N distances but central angles differing substantially from 90 and 180°. Both ligands are basically featureless from a geometric point of view, with torsion angles in both coordinated tetrathionate groups suggesting a trend linking metal size (covalent radius) and ligand `openness'. Packing is directed by (C—H)aromaticO bridges and π–π offset stacked interactions defining chains along [001], further linked by weaker (C—H)methylO bridges, some of them mediated by the dimethylformamide solvent molecules.
O bridges and π–π offset stacked interactions defining chains along [001], further linked by weaker (C—H)methylO bridges, some of them mediated by the dimethylformamide solvent molecules.organic compounds
The natural title compound, C11H12O4, extracted from the Chilean native tree Aristotelia chilensis (Maqui), is a polymorph of the synthetic E form reported by Xia, Hu & Rao [Acta Cryst. (2004), E60, o913-o914]. Both rotational conformers are identical from a metrical point of view, and only differ in the orientation of the 3,4-dihydroxyphenyl ring with respect to the rest of the molecule, which leads to completely different crystal structure arrangements and packing efficiencies. The reasons behind both reside in the different hydrogen-bonding interactions.
metal-organic compounds
The coordinating properties of the novel ligand 4'-[4-(pyrimidin-5-yl)phenyl]-2,2':6',2''-terpyridine were tested by reacting it with cadmium(II) nitrate. The resulting title complex presents a supramolecular motif dominated by a significant number of - interactions involving all the aromatic rings.
- interactions involving all the aromatic rings.metal-organic compounds
The three title transition-metal complexes display a distorted 4+2 octahedral environment for the cation, with the macrocycle defining the base. The effect of the different axial ligands in the intra- and intermolecular hydrogen bonding is discussed.
organic compounds
In the title compound, there are two independent molecules forming hydrogen- and π-bonded dimeric entities with a noticeable noncrystallographic C2 symmetry. Dimers are linked by medium-strength type-I C—FF—C interactions, forming elongated tetramers.
F—C interactions, forming elongated tetramers.metal-organic compounds
In a mononuclear NdIII complex, three chelating 2,4-dioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate ligands and three aqua ligands build up a distorted monocapped square antiprism around the cation. A complex hydrogen-bonding network results in a densely packed structure (packing index = 77.7%)
organic compounds
A polymorphic form of the title indole alkaloid was extracted from Aristotelia chilensis. Its crystal structure is described and compared with a previously reported polymorph.



