Buy article online - an online subscription or single-article purchase is required to access this article.
Crystals of the title compound, (CH
6N
3)
3[H
6CoMo
6O
24]·4H
2O, containing the well known B-type Anderson-Evans heteropolyoxometalate structure, were obtained by recrystallization of powder (CH
6N
3)
3[H
6CoMo
6O
24]·
nH
2O. The anion has a Co
III atom at an inversion center and has approximate
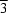
symmetry, with Co-O bond lengths in the range 1.905 (3)-1.918 (3) Å and Mo-O bond lengths in the ranges 1.704 (3)-1.722 (3) (O
t), 1.915 (3)-1.949 (3) (O
b), and 2.266 (3)-2.312 (3) Å (O
c), respectively.
Supporting information
CCDC reference: 214783
Key indicators
- Single-crystal X-ray study
- T = 298 K
- Mean
![[sigma]](//journals.iucr.org/logos/entities/sigma_rmgif.gif) (N-C) = 0.009 Å
(N-C) = 0.009 Å
- H-atom completeness 57%
- Disorder in solvent or counterion
- R factor = 0.031
- wR factor = 0.084
- Data-to-parameter ratio = 15.7
checkCIF results
No syntax errors found
ADDSYM reports no extra symmetry
 Alert Level C:
Alert Level C:
PLAT_302 Alert C Anion/Solvent Disorder ....................... 23.00 Perc.
PLAT_420 Alert C D-H Without Acceptor N2 - H2A ?
General Notes
FORMU_01 There is a discrepancy between the atom counts in the
_chemical_formula_sum and the formula from the _atom_site* data.
Atom count from _chemical_formula_sum:C3 H32 Co1 Mo6 N9 O28
Atom count from the _atom_site data: C3 H18 Co1 Mo6 N9 O28
ABSTM_02 The ratio of expected to reported Tmax/Tmin(RR) is > 1.10
Tmin and Tmax reported: 0.603 0.678
Tmin and Tmax expected: 0.423 0.570
RR = 1.199
Please check that your absorption correction is appropriate.
CELLZ_01
From the CIF: _cell_formula_units_Z 1
From the CIF: _chemical_formula_sum C3 H32 Co Mo6 N9 O28
TEST: Compare cell contents of formula and atom_site data
atom Z*formula cif sites diff
C 3.00 3.00 0.00
H 32.00 18.00 14.00
Co 1.00 1.00 0.00
Mo 6.00 6.00 0.00
N 9.00 9.00 0.00
O 28.00 28.00 0.00
Difference between formula and atom_site contents detected.
WARNING: H atoms missing from atom site list. Is this intentional?
CHEMW_03
From the CIF: _cell_formula_units_Z 1
From the CIF: _chemical_formula_weight 1276.95
TEST: Calculate formula weight from _atom_site_*
atom mass num sum
C 12.01 3.00 36.03
H 1.01 18.00 18.14
N 14.01 9.00 126.06
Mo 95.94 6.00 575.64
O 16.00 28.00 447.97
Co 58.93 1.00 58.93
Calculated formula weight 1262.79
The ratio of given/expected molecular weight as calculated
from the _atom_site* data lies outside
the range 0.99 <> 1.01
0 Alert Level A = Potentially serious problem
0 Alert Level B = Potential problem
2 Alert Level C = Please check
A blue powder of (I) was obtained by adding a small excess stoichiometric amount of CH6N3Cl to a K3[H6CoMo6O24].nH2O solution (Lee et al., 2001). Single crystals of (I) were obtained by recrystallizing the powdered crude sample from a boiling aqueous solution.
The guanidiniun ion that lies on an inversion center was fixed on (1/2, 1/2, 0) and refined under the restraint of FLAT, SAME conditions, and H atoms were generated by PART. All H atoms were placed in calculated positions with N—H distances of 0.86 Å and H—N—H angles of 120°. They were included in the refinement in riding-motion approximation with, Uiso = 1.2Ueq(N). The highest peak in the difference map is 0.43 Å from N5, and the largest hole is 0.42 Å from N5.
Data collection: STADI4 (Stoe & Cie, 1996); cell refinement: STADI4; data reduction: X-RED (Stoe & Cie, 1996); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEP-3 (Farrugia, 1997) and DIAMOND (Brandenburg, 1998).
Crystal data top
| (CH6N3)3[H6CoMo6O24]·4H2O | Z = 1 |
| Mr = 1276.95 | F(000) = 616 |
| Triclinic, P1 | Dx = 2.707 Mg m−3 |
| Hall symbol: -p_1 | Mo Kα radiation, λ = 0.7107 Å |
| a = 8.821 (1) Å | Cell parameters from 27 reflections |
| b = 11.603 (3) Å | θ = 9.5–10.4° |
| c = 8.013 (1) Å | µ = 2.96 mm−1 |
| α = 93.93 (3)° | T = 298 K |
| β = 105.95 (1)° | Polyhedron, blue |
| γ = 83.77 (1)° | 0.38 × 0.25 × 0.19 mm |
| V = 783.3 (2) Å3 | |
Data collection top
Stoe Stadi-4
diffractometer | 3309 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.000 |
| Graphite monochromator | θmax = 27.5°, θmin = 1.8° |
| ω/2–θ scans | h = −11→11 |
Absorption correction: numerical
(X-SHAPE; Stoe & Cie, 1996) | k = −15→15 |
| Tmin = 0.603, Tmax = 0.678 | l = 0→10 |
| 3588 measured reflections | 3 standard reflections every 60 min |
| 3588 independent reflections | intensity decay: 2.0% |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.031 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.084 | H-atom parameters constrained |
| S = 1.15 | w = 1/[σ2(Fo2) + (0.0366P)2 + 2.8997P]
where P = (Fo2 + 2Fc2)/3 |
| 3588 reflections | (Δ/σ)max = 0.001 |
| 229 parameters | Δρmax = 1.42 e Å−3 |
| 7 restraints | Δρmin = −1.64 e Å−3 |
Crystal data top
| (CH6N3)3[H6CoMo6O24]·4H2O | γ = 83.77 (1)° |
| Mr = 1276.95 | V = 783.3 (2) Å3 |
| Triclinic, P1 | Z = 1 |
| a = 8.821 (1) Å | Mo Kα radiation |
| b = 11.603 (3) Å | µ = 2.96 mm−1 |
| c = 8.013 (1) Å | T = 298 K |
| α = 93.93 (3)° | 0.38 × 0.25 × 0.19 mm |
| β = 105.95 (1)° | |
Data collection top
Stoe Stadi-4
diffractometer | 3309 reflections with I > 2σ(I) |
Absorption correction: numerical
(X-SHAPE; Stoe & Cie, 1996) | Rint = 0.000 |
| Tmin = 0.603, Tmax = 0.678 | 3 standard reflections every 60 min |
| 3588 measured reflections | intensity decay: 2.0% |
| 3588 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.031 | 7 restraints |
| wR(F2) = 0.084 | H-atom parameters constrained |
| S = 1.15 | Δρmax = 1.42 e Å−3 |
| 3588 reflections | Δρmin = −1.64 e Å−3 |
| 229 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | Occ. (<1) |
| Co | 0.5000 | 0.0000 | 0.5000 | 0.01253 (15) | |
| Mo1 | 0.54492 (4) | 0.27422 (3) | 0.45785 (5) | 0.01909 (10) | |
| Mo2 | 0.75405 (4) | 0.13827 (3) | 0.81265 (5) | 0.01957 (10) | |
| Mo3 | 0.69975 (4) | −0.13468 (3) | 0.86017 (4) | 0.01856 (10) | |
| Oc1 | 0.3702 (3) | 0.1413 (2) | 0.4394 (4) | 0.0160 (5) | |
| Oc2 | 0.6723 (3) | 0.0911 (2) | 0.5200 (4) | 0.0167 (6) | |
| Oc3 | 0.5409 (3) | 0.0282 (2) | 0.7463 (3) | 0.0150 (5) | |
| Ob4 | 0.4941 (4) | 0.2072 (3) | 0.2236 (4) | 0.0223 (6) | |
| Ob5 | 0.5889 (4) | 0.2565 (3) | 0.7037 (4) | 0.0229 (6) | |
| Ob6 | 0.8349 (4) | −0.0218 (3) | 0.8280 (4) | 0.0235 (6) | |
| Ot7 | 0.4006 (5) | 0.3876 (3) | 0.4360 (5) | 0.0333 (8) | |
| Ot8 | 0.7122 (5) | 0.3319 (3) | 0.4465 (5) | 0.0341 (8) | |
| Ot9 | 0.9163 (4) | 0.1958 (3) | 0.7878 (5) | 0.0375 (9) | |
| Ot10 | 0.7456 (4) | 0.1745 (3) | 1.0201 (4) | 0.0315 (8) | |
| Ot11 | 0.6861 (4) | −0.0991 (3) | 1.0681 (4) | 0.0278 (7) | |
| Ot12 | 0.8312 (4) | −0.2549 (3) | 0.8737 (5) | 0.0329 (8) | |
| C1 | 0.9309 (7) | 0.4957 (5) | 0.2659 (8) | 0.0387 (12) | |
| N1 | 0.8336 (7) | 0.5577 (4) | 0.3460 (8) | 0.0509 (13) | |
| H1a | 0.8386 | 0.6313 | 0.3637 | 0.061* | |
| H1b | 0.7657 | 0.5241 | 0.3801 | 0.061* | |
| N2 | 1.0340 (8) | 0.5444 (5) | 0.2136 (9) | 0.0632 (17) | |
| H2a | 1.0949 | 0.5035 | 0.1603 | 0.076* | |
| H2b | 1.0416 | 0.6177 | 0.2320 | 0.076* | |
| N3 | 0.9172 (7) | 0.3831 (4) | 0.2390 (9) | 0.0593 (17) | |
| H3a | 0.9773 | 0.3410 | 0.1859 | 0.071* | |
| H3b | 0.8482 | 0.3520 | 0.2746 | 0.071* | |
| C2 | 0.5000 | 0.5000 | 0.0000 | 0.091 (6) | |
| N4 | 0.4269 (15) | 0.5214 (8) | 0.1453 (12) | 0.061 (4) | 0.50 |
| H4a | 0.3976 | 0.5912 | 0.1746 | 0.073* | 0.50 |
| H4b | 0.4136 | 0.4641 | 0.2008 | 0.073* | 0.50 |
| N5 | 0.5239 (14) | 0.5628 (10) | −0.0806 (15) | 0.079 (5) | 0.50 |
| H5a | 0.5543 | 0.5400 | −0.1716 | 0.095* | 0.50 |
| H5b | 0.5126 | 0.6357 | −0.0537 | 0.095* | 0.50 |
| N6 | 0.5084 (13) | 0.3763 (7) | −0.0123 (13) | 0.045 (3) | 0.50 |
| H6a | 0.5372 | 0.3391 | −0.0968 | 0.054* | 0.50 |
| H6b | 0.4848 | 0.3390 | 0.0647 | 0.054* | 0.50 |
| Ow1 | −0.0959 (4) | 0.0823 (4) | 0.3372 (5) | 0.0389 (9) | |
| Ow2 | 0.1414 (5) | 0.1924 (3) | 0.5934 (5) | 0.0379 (9) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Co | 0.0136 (3) | 0.0128 (3) | 0.0127 (3) | −0.0021 (3) | 0.0057 (3) | 0.0004 (3) |
| Mo1 | 0.0261 (2) | 0.01388 (17) | 0.02034 (19) | −0.00479 (13) | 0.01006 (14) | 0.00137 (13) |
| Mo2 | 0.02118 (19) | 0.02327 (19) | 0.01604 (18) | −0.00880 (14) | 0.00616 (13) | −0.00232 (13) |
| Mo3 | 0.02037 (18) | 0.01963 (18) | 0.01537 (17) | 0.00139 (13) | 0.00518 (13) | 0.00228 (13) |
| Oc1 | 0.0189 (13) | 0.0145 (13) | 0.0167 (13) | −0.0003 (11) | 0.0085 (11) | 0.0022 (10) |
| Oc2 | 0.0189 (14) | 0.0161 (13) | 0.0175 (13) | −0.0049 (11) | 0.0078 (11) | 0.0001 (11) |
| Oc3 | 0.0156 (13) | 0.0173 (13) | 0.0135 (13) | −0.0035 (10) | 0.0058 (10) | −0.0001 (10) |
| Ob4 | 0.0301 (16) | 0.0220 (15) | 0.0183 (14) | −0.0063 (12) | 0.0105 (12) | 0.0035 (11) |
| Ob5 | 0.0329 (17) | 0.0179 (14) | 0.0204 (15) | −0.0045 (12) | 0.0112 (13) | −0.0026 (11) |
| Ob6 | 0.0170 (14) | 0.0288 (16) | 0.0240 (15) | −0.0032 (12) | 0.0039 (12) | 0.0013 (12) |
| Ot7 | 0.044 (2) | 0.0196 (16) | 0.038 (2) | 0.0038 (14) | 0.0159 (16) | 0.0031 (14) |
| Ot8 | 0.041 (2) | 0.0322 (18) | 0.0355 (19) | −0.0175 (16) | 0.0160 (16) | 0.0011 (15) |
| Ot9 | 0.0329 (19) | 0.048 (2) | 0.035 (2) | −0.0192 (17) | 0.0100 (16) | −0.0006 (17) |
| Ot10 | 0.042 (2) | 0.0348 (19) | 0.0182 (16) | −0.0069 (15) | 0.0092 (14) | −0.0034 (13) |
| Ot11 | 0.0315 (17) | 0.0322 (18) | 0.0197 (15) | −0.0021 (14) | 0.0070 (13) | 0.0011 (13) |
| Ot12 | 0.0308 (18) | 0.0296 (18) | 0.0344 (19) | 0.0084 (14) | 0.0064 (15) | 0.0028 (14) |
| C1 | 0.041 (3) | 0.034 (3) | 0.048 (3) | −0.009 (2) | 0.023 (3) | −0.007 (2) |
| N1 | 0.057 (3) | 0.035 (3) | 0.071 (4) | −0.002 (2) | 0.038 (3) | −0.009 (2) |
| N2 | 0.076 (4) | 0.049 (3) | 0.086 (5) | −0.029 (3) | 0.052 (4) | −0.011 (3) |
| N3 | 0.062 (4) | 0.034 (3) | 0.103 (5) | −0.014 (2) | 0.059 (4) | −0.016 (3) |
| C2 | 0.141 (13) | 0.056 (7) | 0.033 (5) | 0.053 (8) | −0.022 (6) | −0.010 (5) |
| N4 | 0.137 (12) | 0.021 (5) | 0.026 (5) | 0.003 (6) | 0.027 (6) | 0.003 (4) |
| N5 | 0.153 (16) | 0.036 (7) | 0.049 (8) | −0.012 (8) | 0.027 (9) | 0.005 (6) |
| N6 | 0.079 (8) | 0.024 (4) | 0.045 (6) | 0.011 (4) | 0.044 (6) | 0.011 (4) |
| Ow1 | 0.037 (2) | 0.048 (2) | 0.0308 (19) | 0.0020 (17) | 0.0103 (16) | 0.0044 (17) |
| Ow2 | 0.039 (2) | 0.040 (2) | 0.043 (2) | −0.0017 (17) | 0.0257 (17) | 0.0029 (17) |
Geometric parameters (Å, º) top
| Mo1—Mo2 | 3.328 (1) | Mo3—Ot12 | 1.704 (3) |
| Mo2—Mo3 | 3.317 (1) | Mo3—Ot11 | 1.722 (3) |
| Mo1—Mo3i | 3.3081 (9) | Mo3—Ob4i | 1.920 (3) |
| Co—Mo1 | 3.2999 (9) | Mo3—Ob6 | 1.938 (3) |
| Co—Mo2 | 3.3191 (9) | Mo3—Oc1i | 2.307 (3) |
| Co—Mo3 | 3.3319 (9) | Mo3—Oc3 | 2.308 (3) |
| Co—Oc1 | 1.905 (3) | Oc3—Ot11ii | 2.831 (4) |
| Co—Oc2 | 1.909 (3) | C1—N2 | 1.295 (7) |
| Co—Oc3 | 1.918 (3) | C1—N3 | 1.323 (7) |
| Mo1—Ot7 | 1.711 (3) | C1—N1 | 1.331 (7) |
| Mo1—Ot8 | 1.712 (3) | C2—N5 | 1.08 (1) |
| Mo1—Ob5 | 1.921 (3) | C2—N6 | 1.427 (8) |
| Mo1—Ob4 | 1.934 (3) | C2—N4 | 1.473 (9) |
| Mo1—Oc1 | 2.266 (3) | Ow1—Oc2iii | 2.814 (5) |
| Mo1—Oc2 | 2.312 (3) | Ow1—Ot10iv | 2.765 (5) |
| Mo2—Ot10 | 1.706 (3) | Ow1—Ob6i | 2.956 (5) |
| Mo2—Ot9 | 1.707 (3) | Ow1—Ow2 | 2.843 (6) |
| Mo2—Ob6 | 1.915 (3) | Ow2—Oc1 | 2.638 (4) |
| Mo2—Ob5 | 1.949 (3) | Ow2—Ot9iii | 2.837 (5) |
| Mo2—Oc2 | 2.302 (3) | Ow2—Ot11ii | 2.937 (5) |
| Mo2—Oc3 | 2.308 (3) | Ow2—N1v | 2.930 (7) |
| | | |
| Mo3—Mo2—Mo1 | 119.78 (3) | Ot10—Mo2—Oc3 | 95.06 (14) |
| Mo3i—Mo1—Mo2 | 120.63 (2) | Ot9—Mo2—Oc3 | 158.18 (15) |
| Oc1—Co—Oc2i | 95.10 (12) | Ob6—Mo2—Oc3 | 72.12 (12) |
| Oc1—Co—Oc2 | 84.90 (12) | Ob5—Mo2—Oc3 | 81.28 (12) |
| Oc2i—Co—Oc2 | 180.0 | Oc2—Mo2—Oc3 | 68.22 (10) |
| Oc1—Co—Oc3 | 95.56 (12) | Ot12—Mo3—Ot11 | 106.57 (17) |
| Oc1i—Co—Oc3 | 84.44 (12) | Ot12—Mo3—Ob6 | 98.30 (16) |
| Oc2i—Co—Oc3 | 95.00 (12) | Ot11—Mo3—Ob6 | 101.96 (15) |
| Oc2—Co—Oc3 | 85.00 (12) | Ot12—Mo3—Oc3 | 159.32 (14) |
| Ot7—Mo1—Ot8 | 106.05 (18) | Ot11—Mo3—Oc3 | 93.41 (13) |
| Ot7—Mo1—Ob5 | 98.69 (16) | Ob6—Mo3—Oc3 | 71.75 (11) |
| Ot8—Mo1—Ob5 | 101.04 (16) | N2—C1—N3 | 121.1 (5) |
| Ot7—Mo1—Ob4 | 100.43 (16) | N2—C1—N1 | 121.1 (5) |
| Ot8—Mo1—Ob4 | 95.84 (16) | N3—C1—N1 | 117.9 (5) |
| Ob5—Mo1—Ob4 | 149.82 (13) | N5—C2—N6 | 132.1 (8) |
| Ot7—Mo1—Oc1 | 92.65 (15) | N5—C2—N4 | 128.1 (8) |
| Ot8—Mo1—Oc1 | 159.47 (15) | N6—C2—N4 | 99.4 (5) |
| Ob5—Mo1—Oc1 | 84.02 (12) | Ot10iv—Ow1—Oc2iii | 104.60 (15) |
| Ob4—Mo1—Oc1 | 72.01 (11) | Ot10iv—Ow1—Ow2 | 124.06 (18) |
| Ot7—Mo1—Oc2 | 159.33 (14) | Oc2iii—Ow1—Ow2 | 98.87 (15) |
| Ot8—Mo1—Oc2 | 94.01 (15) | Ot10iv—Ow1—Ob6i | 84.11 (14) |
| Ob5—Mo1—Oc2 | 72.02 (12) | Oc2iii—Ow1—Ob6i | 167.65 (18) |
| Ob4—Mo1—Oc2 | 82.07 (12) | Ow2—Ow1—Ob6i | 82.89 (14) |
| Oc1—Mo1—Oc2 | 68.41 (10) | Oc1—Ow2—Ot9iii | 166.92 (19) |
| Ot10—Mo2—Ot9 | 106.45 (18) | Oc1—Ow2—Ow1 | 96.44 (16) |
| Ot10—Mo2—Ob6 | 102.30 (16) | Ot9iii—Ow2—Ow1 | 82.23 (15) |
| Ot9—Mo2—Ob6 | 99.15 (17) | Oc1—Ow2—N1v | 98.81 (17) |
| Ot10—Mo2—Ob5 | 95.67 (15) | Ot9iii—Ow2—N1v | 92.29 (17) |
| Ot9—Mo2—Ob5 | 99.79 (17) | Ow1—Ow2—N1v | 126.9 (2) |
| OB6—Mo2—Ob5 | 148.91 (13) | Oc1—Ow2—Ot11ii | 96.96 (15) |
| Ot10—Mo2—Oc2 | 160.06 (15) | Ot9iii—Ow2—Ot11ii | 73.68 (14) |
| Ot9—Mo2—Oc2 | 91.24 (15) | Ow1—Ow2—Ot11ii | 125.36 (17) |
| Ob6—Mo2—Oc2 | 83.38 (12) | N1v—Ow2—Ot11ii | 102.79 (19) |
| Ob5—Mo2—Oc2 | 71.80 (11) | | |
| Symmetry codes: (i) −x+1, −y, −z+1; (ii) −x+1, −y, −z+2; (iii) x−1, y, z; (iv) x−1, y, z−1; (v) −x+1, −y+1, −z+1. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1b···Ot7v | 0.86 | 2.46 | 3.044 (6) | 126 |
| N2—H2b···Ot9vi | 0.86 | 2.26 | 3.094 (7) | 164 |
| N3—H3a···Ot12vii | 0.86 | 2.02 | 2.855 (6) | 163 |
| N3—H3b···Ot8 | 0.86 | 2.10 | 2.900 (6) | 155 |
| N4—H4a···Ob5v | 0.86 | 1.96 | 2.78 (1) | 159 |
| N4—H4b···Ot7 | 0.86 | 2.17 | 2.96 (1) | 152 |
| N5—H5b···Ob4viii | 0.86 | 2.34 | 2.95 (1) | 128 |
| N6—H6a···Ob5ix | 0.86 | 1.94 | 2.803 (9) | 177 |
| N6—H6b···Ob4 | 0.86 | 2.04 | 2.852 (9) | 158 |
| N6—H6a···Ot10ix | 0.86 | 2.54 | 2.941 (9) | 110 |
| Symmetry codes: (v) −x+1, −y+1, −z+1; (vi) −x+2, −y+1, −z+1; (vii) −x+2, −y, −z+1; (viii) −x+1, −y+1, −z; (ix) x, y, z−1. |
Experimental details
| Crystal data |
| Chemical formula | (CH6N3)3[H6CoMo6O24]·4H2O |
| Mr | 1276.95 |
| Crystal system, space group | Triclinic, P1 |
| Temperature (K) | 298 |
| a, b, c (Å) | 8.821 (1), 11.603 (3), 8.013 (1) |
| α, β, γ (°) | 93.93 (3), 105.95 (1), 83.77 (1) |
| V (Å3) | 783.3 (2) |
| Z | 1 |
| Radiation type | Mo Kα |
| µ (mm−1) | 2.96 |
| Crystal size (mm) | 0.38 × 0.25 × 0.19 |
| |
| Data collection |
| Diffractometer | Stoe Stadi-4
diffractometer |
| Absorption correction | Numerical
(X-SHAPE; Stoe & Cie, 1996) |
| Tmin, Tmax | 0.603, 0.678 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 3588, 3588, 3309 |
| Rint | 0.000 |
| (sin θ/λ)max (Å−1) | 0.649 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.031, 0.084, 1.15 |
| No. of reflections | 3588 |
| No. of parameters | 229 |
| No. of restraints | 7 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 1.42, −1.64 |
Selected bond lengths (Å) top| Mo1—Mo2 | 3.328 (1) | Mo3—Ot12 | 1.704 (3) |
| Mo2—Mo3 | 3.317 (1) | Mo3—Ot11 | 1.722 (3) |
| Mo1—Mo3i | 3.3081 (9) | Mo3—Ob4i | 1.920 (3) |
| Co—Mo1 | 3.2999 (9) | Mo3—Ob6 | 1.938 (3) |
| Co—Mo2 | 3.3191 (9) | Mo3—Oc1i | 2.307 (3) |
| Co—Mo3 | 3.3319 (9) | Mo3—Oc3 | 2.308 (3) |
| Co—Oc1 | 1.905 (3) | Oc3—Ot11ii | 2.831 (4) |
| Co—Oc2 | 1.909 (3) | C1—N2 | 1.295 (7) |
| Co—Oc3 | 1.918 (3) | C1—N3 | 1.323 (7) |
| Mo1—Ot7 | 1.711 (3) | C1—N1 | 1.331 (7) |
| Mo1—Ot8 | 1.712 (3) | C2—N5 | 1.08 (1) |
| Mo1—Ob5 | 1.921 (3) | C2—N6 | 1.427 (8) |
| Mo1—Ob4 | 1.934 (3) | C2—N4 | 1.473 (9) |
| Mo1—Oc1 | 2.266 (3) | Ow1—Oc2iii | 2.814 (5) |
| Mo1—Oc2 | 2.312 (3) | Ow1—Ot10iv | 2.765 (5) |
| Mo2—Ot10 | 1.706 (3) | Ow1—Ob6i | 2.956 (5) |
| Mo2—Ot9 | 1.707 (3) | Ow1—Ow2 | 2.843 (6) |
| Mo2—Ob6 | 1.915 (3) | Ow2—Oc1 | 2.638 (4) |
| Mo2—Ob5 | 1.949 (3) | Ow2—Ot9iii | 2.837 (5) |
| Mo2—Oc2 | 2.302 (3) | Ow2—Ot11ii | 2.937 (5) |
| Mo2—Oc3 | 2.308 (3) | Ow2—N1v | 2.930 (7) |
| Symmetry codes: (i) −x+1, −y, −z+1; (ii) −x+1, −y, −z+2; (iii) x−1, y, z; (iv) x−1, y, z−1; (v) −x+1, −y+1, −z+1. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1b···Ot7v | 0.86 | 2.46 | 3.044 (6) | 126 |
| N2—H2b···Ot9vi | 0.86 | 2.26 | 3.094 (7) | 164 |
| N3—H3a···Ot12vii | 0.86 | 2.02 | 2.855 (6) | 163 |
| N3—H3b···Ot8 | 0.86 | 2.10 | 2.900 (6) | 155 |
| N4—H4a···Ob5v | 0.86 | 1.96 | 2.78 (1) | 159 |
| N4—H4b···Ot7 | 0.86 | 2.17 | 2.96 (1) | 152 |
| N5—H5b···Ob4viii | 0.86 | 2.34 | 2.95 (1) | 128 |
| N6—H6a···Ob5ix | 0.86 | 1.94 | 2.803 (9) | 177 |
| N6—H6b···Ob4 | 0.86 | 2.04 | 2.852 (9) | 158 |
| N6—H6a···Ot10ix | 0.86 | 2.54 | 2.941 (9) | 110 |
| Symmetry codes: (v) −x+1, −y+1, −z+1; (vi) −x+2, −y+1, −z+1; (vii) −x+2, −y, −z+1; (viii) −x+1, −y+1, −z; (ix) x, y, z−1. |

 Subscribe to Acta Crystallographica Section E: Crystallographic Communications
Subscribe to Acta Crystallographica Section E: Crystallographic Communications
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu metal-organic compounds
metal-organic compounds














![[Figure 1]](http://journals.iucr.org/e/issues/2003/06/00/ww6068/ww6068fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/e/issues/2003/06/00/ww6068/ww6068fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/e/issues/2003/06/00/ww6068/ww6068fig3thm.gif)




We have recently reported the crystal structures of two guanidinium salts of polyoxometalates, viz. (CH6N3)8[PtW6O24] (Lee, Jang et al., 2003). and (CH6N3)8[SiPt2W10O40]·6H2O (Lee, Joo et al., 2003). Also, (CH6N3)4[GeMo12O40] (Strandberg & Hedman, 1982), (CH6N3)2[MoO4] (Ozeki et al., 1987), (CH6N3)4[SiMo12O40]·H2O (Ichida et al., 1980), (CH6N3)4[As2Mo18O62]·9H2O (Ichida & Sasaki, 1983), and (CH6N3)6[V10O28]·6H2O (Wang et al., 1993) have been reported by others. Good crystals of guanidinium polyoxometalates can not always be obtained, but sometimes very stable single crystals are obtained by recrystallization. And, these salts were shown to be arresting crystal packing by hydrogen bonds and thermal behavior of crystalline water molecules.
Several heteropolyoxometalates containing the [H6CoMo6O24]3− polyanion, such as (18-crown-6.K+)2·K[H6CoMo6O24].12H2O (Nagano et al., 1990), [Ga(H2O)6]3[H6CoMo6O24].10H2O (Panneerselvam et al., 1996), Na3[H6CoMo6O24]·8H2O (Nolan et al., 1998), K3[H6CoMo6O24]·KNO3·4H2O (Lee & Joo, 2000), and K3[H6CoMo6O24]·4H2O (Lee et al., 2001), have been reported. The [H6CoMo6O24]3− anion is a typical B-type Anderson–Evans heteropolyanion structure (Anderson, 1937; Evans, 1948; Tsigdinos, 1978). Detailed discussions related to the position of six H atoms in the [H6CoMo6O24]3− have been given by Nolan et al. (1998). Six non-acidic H atoms are bound to six central Oc atoms surrounding the CoIII atom. As expected, they were not substituted by guanidinium ion. The crystal structure of (I) is a good example of the packing and the hydrogen bonding found in the B-type Anderson–Evans heteropolyoxometalate structure and thermal behavior of crystalline water molecules. Here we report on the crystal structure of (I), in which the replaceable cations were completely exchanged by guanidinium ions.
Fig. 1 shows the structure of the [H6CoMo6O24]3− polyanion. The labeling of the O atoms in the polyanion is the same as the labeling in the previous structure (Lee et al., 2001). The distance ranges of Co—Mo and Mo—Mo are 3.2999 (9)–3.3319 (9) and 3.3081 (9)–3.328 (1) Å, respectively. The bond-length range of Co—O is 1.905 (3)–1.918 (3) Å, and the three types of Mo—O bond-length ranges are 1.704 (3)–1.722 (3) (Ot), 1.915 (3)–1.949 (3) (Ob), and 2.266 (3)–2.312 (3) Å (Oc). The bond lengths and angles of (I) have shown a similar trend to what is found in the literatures, and the guanidinium ions also agree well with those in CH6N3Cl (Hass et al., 1965) and CH6N3(NO3) (Katrusiak& Szafrański, 1994). There are one and a half crystallographically independent guanidinium (CH6N3)+ ions in the asymmetric unit, one of which lies on an inversion center.
Fig. 2 shows the hydrogen-bonding interaction of (CH6N3)+ ions with O atoms of the anions and crystalline water molecules. The list of all probable hydrogen bonds with N···O and O···O distances within 3.1 Å is given in Tables 1 and 2.An iInter-anion hydrogen bond is formed by Oc3—H···Ot11 [2.831 (4) Å]. All O atoms in the polyanion except Oc3 form strong N—H····O and O—H····O hydrogen bonds, with all N atoms of the guanidinium ions and all the crystalline water molecules. Each crystalline water molecule forms strong hydrogen bonds. As a result, the degradation temperature was very high (543 K) and those were decomposed with (CH6N3)+ ions. All probable N—Opoly anion hydrogen-bond distances fall into the range 2.78–3.10 Å, with the range of N—H···O angles varying between 119 and 177°. The crystal structure of (I) is stabilized by the formation of extensive N—H···O and O—H···O hydrogen-bond interactions with the guanidinium ions and the waters of crystallization. Fig. 3 shows the unit-cell packing and probable hydrogen-bonding interactions in the lattice.