

 journal menu
journal menu inorganic compounds
inorganic compounds









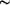 0.60 Å about the 31 or 32 axes. Normal probability analysis reveals systematic error in seven or more of the earlier atomic coordinates.
0.60 Å about the 31 or 32 axes. Normal probability analysis reveals systematic error in seven or more of the earlier atomic coordinates.Supporting information
 | Crystallographic Information File (CIF) https://doi.org/10.1107/S0108270104026368/ta1472sup1.cif |
 | Structure factor file (CIF format) https://doi.org/10.1107/S0108270104026368/ta1472Isup2.hkl |
| Structure factor file (CIF format) https://doi.org/10.1107/S0108270104026368/ta1472IIsup3.hkl |
 | Portable Document Format (PDF) file https://doi.org/10.1107/S0108270104026368/ta1472sup4.pdf |
Averbuch-Pouchot et al. (1978) prepared the title chromatoarsenate by the reaction K2Cr2O7 + H3AsO4 → K2HCr2AsO10 + H2O. Substitution of either 1/2[As2O5.xH2O] with x ≈ 3 or KH2AsO4 for H3AsO4 also yields K2HCr2AsO10 (CAS Registry No. 69107–38-6). Initial crystal growth in either reaction using 99.9% pure starting products with 0.1 mol of each reactant dissolved in about 10 mol H2O, on heating to boiling for several minutes and then cooling naturally to room temperature, commonly gives two crops of strong reddish-orange [ISCC-NBS Color (Supplement to NBS Circular 553). A complete list with RGB values is given at https://swiss.csail.mit.edu/jaffer/Color/nbs-iscc-rgb.pdf Link broken and also at https://tx4.us/nbs-iscc.htm] crystals (Weakley et al., 2004). Two crystals from first-growth crops were selected for the present study. The solubility at 298 K was determined as 207 g l−1.
The general structure solution made use of a SIR92 electron-density map (Altomare et al., 1994). The Flack (1983) parameter shows that the space group for crystal 1 is primarily P31 but with a small enantiomorphic component, whereas the space group for crystal 2 does not differ significantly from P32. The H atom could not be located directly with confidence in either crystal but was later inferred (see Comment).
For both compounds, data collection: CAD-4-PC Software (Enraf-Nonius, 1993); cell refinement: CAD-4-PC Software; data reduction: TEXSAN (Molecular Structure Corporation, 1997); program(s) used to solve structure: TEXSAN; program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: ATOMS (Dowty, 2003).
![[Figure 1]](http://journals.iucr.org/c/issues/2004/12/00/ta1472/ta1472fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2004/12/00/ta1472/ta1472fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2004/12/00/ta1472/ta1472fig3thm.gif)
| Fig. 1(a). The K2HCr2AsO10 structure at 298 K. The O6—H···O5 hydrogen bonds are shown as dashed lines. The labelling of all atoms in the unit cell is given in Supplementary Fig. S1. Fig. 1(b). The HCr2AsO102− ion of crystal 1, showing the interionic hydrogen bond, with displacement ellipsoids drawn at the 50% probability level. The H atom is shown as a sphere and the hydrogen bond as single lines. [Symmetry code: (i) y-x, −x, z − 1/3]. Fig. 2. The K2HCr2AsO10 structure at 298 K, with H atoms shown as small spheres and K atoms as large spheres. The AsO4 tetrahedra are plus-hatched and the CrO4 tetrahedra are cross-hatched. Fig. 3. A normal probability Qexp - Qnorm plot for the atomic coordinates determined for K2HCr2AsO10, with data for crystal 1 plotted versus those for crystal 2. |
| K2[HCr2AsO10] | Dx = 2.779 Mg m−3 |
| Mr = 418.13 | Mo Kα radiation, λ = 0.71073 Å |
| Trigonal, P31 | Cell parameters from 25 reflections |
| Hall symbol: P 31 | θ = 14.1–16.4° |
| a = 7.6931 (8) Å | µ = 6.33 mm−1 |
| c = 14.623 (3) Å | T = 296 K |
| V = 749.50 (19) Å3 | Hexagonal prism, intense red-orange |
| Z = 3 | 0.40 × 0.22 × 0.18 mm |
| F(000) = 600 |
| Enraf-Nonius CAD-4 diffractometer | 2040 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.019 |
| Graphite monochromator | θmax = 30.0°, θmin = 3.1° |
| ω/2θ scans | h = −8→10 |
| Absorption correction: analytical (de Meulenaer & Tompa, 1965) | k = 0→10 |
| Tmin = 0.205, Tmax = 0.320 | l = −20→20 |
| 2063 measured reflections | 3 standard reflections every 300 reflections |
| 2051 independent reflections | intensity decay: 0.0% |
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H atoms treated by a mixture of independent and constrained refinement |
| R[F2 > 2σ(F2)] = 0.050 | w = 1/[σ2(Fo2) + (0.0086P)2 + 7.6027P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.129 | (Δ/σ)max = 0.005 |
| S = 1.29 | Δρmax = 1.31 e Å−3 |
| 2051 reflections | Δρmin = −1.08 e Å−3 |
| 137 parameters | Extinction correction: Zachariasen (1967), Acta Cryst. (1968). A24, p. 213, eq. (3) |
| 1 restraint | Extinction coefficient: 0.0000072 (11) |
| Primary atom site location: structure-invariant direct methods | Absolute structure: Flack (1983), with how many Friedel pairs |
| Secondary atom site location: difference Fourier map | Absolute structure parameter: 0.17 (3) |
| K2[HCr2AsO10] | Z = 3 |
| Mr = 418.13 | Mo Kα radiation |
| Trigonal, P31 | µ = 6.33 mm−1 |
| a = 7.6931 (8) Å | T = 296 K |
| c = 14.623 (3) Å | 0.40 × 0.22 × 0.18 mm |
| V = 749.50 (19) Å3 |
| Enraf-Nonius CAD-4 diffractometer | 2040 reflections with I > 2σ(I) |
| Absorption correction: analytical (de Meulenaer & Tompa, 1965) | Rint = 0.019 |
| Tmin = 0.205, Tmax = 0.320 | 3 standard reflections every 300 reflections |
| 2063 measured reflections | intensity decay: 0.0% |
| 2051 independent reflections |
| R[F2 > 2σ(F2)] = 0.050 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.129 | Δρmax = 1.31 e Å−3 |
| S = 1.29 | Δρmin = −1.08 e Å−3 |
| 2051 reflections | Absolute structure: Flack (1983), with how many Friedel pairs |
| 137 parameters | Absolute structure parameter: 0.17 (3) |
| 1 restraint |
Experimental. The correct polarity in Crystal 1 was ascertained by refinement in space-groups P31 and P32. It was confirmed by refinement of a Flack parameter; the significantly non-zero value is ascribed to a minor twin component. |
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors (gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R-factors based on ALL data will be even larger. |
| x | y | z | Uiso*/Ueq | ||
| As1 | 0.10826 (13) | −0.02094 (14) | 0.49989 (8) | 0.01945 (18) | |
| Cr1 | 0.0041 (2) | −0.4249 (2) | 0.60617 (10) | 0.0200 (3) | |
| Cr2 | 0.4201 (2) | 0.4191 (2) | 0.43023 (10) | 0.0201 (3) | |
| O1 | −0.1383 (15) | −0.3891 (14) | 0.6749 (7) | 0.039 (2) | |
| O2 | −0.1214 (19) | −0.5908 (15) | 0.5301 (7) | 0.055 (3) | |
| O3 | 0.1440 (15) | −0.4780 (16) | 0.6664 (8) | 0.046 (2) | |
| O4 | 0.1691 (11) | −0.1884 (12) | 0.5449 (6) | 0.0289 (16) | |
| O5 | −0.0079 (14) | −0.0971 (13) | 0.4020 (5) | 0.0361 (19) | |
| O6 | −0.0392 (14) | 0.0180 (15) | 0.5724 (5) | 0.0349 (18) | |
| O7 | 0.3389 (12) | 0.1870 (11) | 0.4962 (6) | 0.0310 (17) | |
| O8 | 0.2329 (16) | 0.4022 (16) | 0.3750 (8) | 0.046 (2) | |
| O9 | 0.5089 (19) | 0.5990 (14) | 0.5027 (6) | 0.047 (3) | |
| O10 | 0.5860 (14) | 0.4415 (15) | 0.3572 (6) | 0.0375 (19) | |
| K1 | −0.0199 (4) | −0.4513 (3) | 0.34633 (17) | 0.0288 (4) | |
| K2 | 0.6027 (4) | −0.0061 (4) | 0.51916 (17) | 0.0292 (4) | |
| H | 0.0256 | −0.0279 | 0.3206 | 0.050* |
| U11 | U22 | U33 | U12 | U13 | U23 | |
| As1 | 0.0201 (4) | 0.0202 (4) | 0.0171 (3) | 0.0094 (4) | −0.0003 (3) | 0.0027 (3) |
| Cr1 | 0.0197 (7) | 0.0170 (6) | 0.0210 (7) | 0.0075 (6) | 0.0006 (5) | 0.0047 (5) |
| Cr2 | 0.0223 (7) | 0.0167 (6) | 0.0211 (6) | 0.0096 (6) | 0.0005 (5) | 0.0011 (5) |
| O1 | 0.040 (5) | 0.035 (5) | 0.048 (5) | 0.023 (4) | 0.016 (4) | 0.012 (4) |
| O2 | 0.072 (8) | 0.030 (5) | 0.031 (4) | 0.002 (5) | −0.004 (4) | −0.005 (3) |
| O3 | 0.034 (5) | 0.046 (6) | 0.060 (6) | 0.022 (4) | −0.002 (4) | 0.020 (5) |
| O4 | 0.022 (3) | 0.030 (4) | 0.038 (4) | 0.015 (3) | 0.007 (3) | 0.016 (3) |
| O5 | 0.043 (5) | 0.039 (5) | 0.013 (3) | 0.010 (4) | −0.007 (3) | −0.002 (3) |
| O6 | 0.050 (5) | 0.049 (5) | 0.021 (4) | 0.036 (5) | −0.004 (3) | −0.007 (3) |
| O7 | 0.020 (3) | 0.017 (3) | 0.042 (4) | −0.002 (3) | −0.006 (3) | 0.011 (3) |
| O8 | 0.045 (5) | 0.048 (6) | 0.052 (6) | 0.029 (5) | −0.003 (4) | 0.008 (4) |
| O9 | 0.082 (8) | 0.028 (4) | 0.025 (4) | 0.024 (5) | 0.002 (4) | −0.007 (3) |
| O10 | 0.037 (5) | 0.047 (5) | 0.030 (4) | 0.023 (4) | 0.005 (3) | 0.005 (3) |
| K1 | 0.0296 (11) | 0.0286 (11) | 0.0291 (10) | 0.0153 (9) | 0.0029 (8) | 0.0010 (8) |
| K2 | 0.0298 (11) | 0.0278 (11) | 0.0278 (10) | 0.0128 (9) | −0.0017 (9) | −0.0010 (8) |
| As1—O5 | 1.633 (7) | K1—O3ii | 2.786 (14) |
| As1—O6 | 1.685 (8) | K1—O5 | 2.801 (10) |
| As1—O7 | 1.695 (7) | K1—O7iii | 3.139 (10) |
| As1—O4 | 1.707 (7) | K1—O8iv | 2.724 (14) |
| Cr1—O3 | 1.594 (9) | K1—O9iii | 2.826 (11) |
| Cr1—O2 | 1.602 (10) | K1—O10v | 2.720 (12) |
| Cr1—O1 | 1.609 (9) | K2—O1ii | 2.828 (11) |
| Cr1—O4 | 1.847 (7) | K2—O2vi | 2.821 (11) |
| Cr2—O8 | 1.599 (10) | K2—O3ii | 3.112 (13) |
| Cr2—O9 | 1.600 (9) | K2—O4 | 2.925 (10) |
| Cr2—O10 | 1.606 (9) | K2—O6vii | 2.778 (12) |
| Cr2—O7 | 1.842 (7) | K2—O7 | 3.075 (10) |
| O5—H | 1.28 | K2—O8viii | 3.030 (13) |
| K1—O1i | 2.742 (10) | K2—O9iv | 2.760 (10) |
| K1—O2 | 2.854 (11) | K2—O10viii | 2.852 (9) |
| O5···O6iii | 2.550 (11) | ||
| O5—As1—O6 | 108.6 (5) | O1—Cr1—O4 | 109.0 (4) |
| O5—As1—O7 | 115.9 (4) | O8—Cr2—O9 | 112.5 (6) |
| O6—As1—O7 | 109.4 (5) | O8—Cr2—O10 | 107.9 (5) |
| O5—As1—O4 | 112.0 (5) | O9—Cr2—O10 | 112.0 (6) |
| O6—As1—O4 | 110.7 (4) | O8—Cr2—O7 | 109.3 (5) |
| O7—As1—O4 | 99.9 (4) | O9—Cr2—O7 | 106.4 (4) |
| O3—Cr1—O2 | 113.2 (7) | O10—Cr2—O7 | 108.6 (5) |
| O3—Cr1—O1 | 107.7 (6) | As1—O4—Cr1 | 128.1 (4) |
| O2—Cr1—O1 | 112.3 (6) | As1—O5—H | 135.1 |
| O3—Cr1—O4 | 107.5 (4) | As1—O7—Cr2 | 128.3 (5) |
| O2—Cr1—O4 | 107.0 (5) |
| Symmetry codes: (i) −x+y, −x−1, z−1/3; (ii) −x+y+1, −x, z−1/3; (iii) −x+y, −x, z−1/3; (iv) x, y−1, z; (v) x−1, y−1, z; (vi) x+1, y+1, z; (vii) x+1, y, z; (viii) −y+1, x−y, z+1/3. |
| K2[HCr2AsO10] | Dx = 2.778 Mg m−3 |
| Mr = 418.13 | Mo Kα radiation, λ = 0.71073 Å |
| Trigonal, P32 | Cell parameters from 25 reflections |
| Hall symbol: P 32 | θ = 13.5–14.9° |
| a = 7.6963 (9) Å | µ = 6.33 mm−1 |
| c = 14.6171 (11) Å | T = 294 K |
| V = 749.82 (14) Å3 | Hexagonal prism, intense-orange |
| Z = 3 | 0.29 × 0.13 × 0.11 mm |
| F(000) = 600 |
| Enraf-Nonius CAD-4 diffractometer | Rint = 0.011 |
| Radiation source: fine-focus sealed tube | θmax = 27.5°, θmin = 3.1° |
| Graphite monochromator | h = −8→8 |
| ω/2θ scans | k = 0→9 |
| 1229 measured reflections | l = −18→18 |
| 1224 independent reflections | 3 standard reflections every 300 reflections |
| 1182 reflections with I > 2σ(I) | intensity decay: 0.5% |
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H atoms treated by a mixture of independent and constrained refinement |
| R[F2 > 2σ(F2)] = 0.021 | w = 1/[σ2(Fo2) + (0.0181P)2 + 0.0502P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.053 | (Δ/σ)max = 0.008 |
| S = 1.08 | Δρmax = 0.46 e Å−3 |
| 1224 reflections | Δρmin = −0.33 e Å−3 |
| 137 parameters | Extinction correction: Zachariasen (1967), Acta Cryst. (1968). A24, p. 213, eq. (3) |
| 1 restraint | Extinction coefficient: 0.0000096 (12) |
| Primary atom site location: structure-invariant direct methods | Absolute structure: Flack (1983), with how many Friedel pairs |
| Secondary atom site location: difference Fourier map | Absolute structure parameter: 0.030 (16) |
| K2[HCr2AsO10] | Z = 3 |
| Mr = 418.13 | Mo Kα radiation |
| Trigonal, P32 | µ = 6.33 mm−1 |
| a = 7.6963 (9) Å | T = 294 K |
| c = 14.6171 (11) Å | 0.29 × 0.13 × 0.11 mm |
| V = 749.82 (14) Å3 |
| Enraf-Nonius CAD-4 diffractometer | Rint = 0.011 |
| 1229 measured reflections | 3 standard reflections every 300 reflections |
| 1224 independent reflections | intensity decay: 0.5% |
| 1182 reflections with I > 2σ(I) |
| R[F2 > 2σ(F2)] = 0.021 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.053 | Δρmax = 0.46 e Å−3 |
| S = 1.08 | Δρmin = −0.33 e Å−3 |
| 1224 reflections | Absolute structure: Flack (1983), with how many Friedel pairs |
| 137 parameters | Absolute structure parameter: 0.030 (16) |
| 1 restraint |
Experimental. The correct polarity in Crystal 2 was ascertained by refinement in both space-group P31 and P32. It was confirmed by refinement of a Flack parameter; the significantly-zero value validates the absence of any enantiomorphic component. |
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors (gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R-factors based on ALL data will be even larger. |
| x | y | z | Uiso*/Ueq | ||
| As1 | 0.12958 (6) | 0.02179 (6) | 0.50004 (5) | 0.01795 (11) | |
| Cr1 | 0.00117 (11) | −0.41910 (11) | 0.43038 (5) | 0.01938 (16) | |
| Cr2 | 0.42899 (11) | 0.42481 (11) | 0.60655 (5) | 0.01996 (16) | |
| O1 | 0.1424 (6) | −0.4423 (7) | 0.3570 (3) | 0.0379 (10) | |
| O2 | −0.0883 (8) | −0.5988 (6) | 0.5024 (3) | 0.0491 (12) | |
| O3 | −0.1692 (6) | −0.4012 (7) | 0.3758 (3) | 0.0421 (10) | |
| O4 | 0.1520 (6) | −0.1879 (6) | 0.4966 (3) | 0.0318 (8) | |
| O5 | 0.0914 (6) | 0.1001 (5) | 0.4024 (2) | 0.0309 (8) | |
| O6 | −0.0582 (5) | −0.0195 (7) | 0.5728 (2) | 0.0330 (9) | |
| O7 | 0.3559 (5) | 0.1869 (5) | 0.5468 (3) | 0.0258 (7) | |
| O8 | 0.6242 (6) | 0.4793 (7) | 0.6657 (4) | 0.0465 (12) | |
| O9 | 0.4665 (9) | 0.5909 (6) | 0.5314 (3) | 0.0515 (13) | |
| O10 | 0.2526 (7) | 0.3923 (7) | 0.6749 (3) | 0.0388 (9) | |
| K1 | 0.43080 (16) | 0.45187 (16) | 0.34636 (8) | 0.0280 (2) | |
| K2 | 0.60914 (17) | 0.00606 (17) | 0.51907 (8) | 0.0285 (2) | |
| H | 0.0256 | −0.0279 | 0.3206 | 0.050* |
| U11 | U22 | U33 | U12 | U13 | U23 | |
| As1 | 0.0195 (2) | 0.0160 (2) | 0.01738 (19) | 0.00821 (18) | −0.00247 (17) | −0.00145 (17) |
| Cr1 | 0.0200 (3) | 0.0152 (3) | 0.0205 (4) | 0.0070 (3) | −0.0008 (3) | −0.0013 (3) |
| Cr2 | 0.0219 (3) | 0.0160 (3) | 0.0216 (4) | 0.0092 (3) | −0.0049 (3) | −0.0050 (3) |
| O1 | 0.039 (2) | 0.048 (2) | 0.036 (2) | 0.029 (2) | 0.0047 (18) | −0.0059 (18) |
| O2 | 0.071 (3) | 0.0216 (19) | 0.030 (2) | 0.004 (2) | 0.001 (2) | 0.0050 (17) |
| O3 | 0.031 (2) | 0.046 (3) | 0.051 (3) | 0.021 (2) | −0.0144 (19) | −0.009 (2) |
| O4 | 0.036 (2) | 0.0216 (16) | 0.040 (2) | 0.0164 (16) | −0.0169 (17) | −0.0068 (15) |
| O5 | 0.042 (2) | 0.0284 (19) | 0.0218 (17) | 0.0175 (17) | −0.0046 (15) | 0.0024 (14) |
| O6 | 0.0225 (17) | 0.052 (3) | 0.0227 (18) | 0.0175 (17) | 0.0042 (14) | 0.0040 (17) |
| O7 | 0.0196 (16) | 0.0216 (16) | 0.0343 (19) | 0.0089 (13) | −0.0048 (14) | −0.0153 (14) |
| O8 | 0.031 (2) | 0.050 (3) | 0.059 (3) | 0.021 (2) | −0.023 (2) | −0.028 (2) |
| O9 | 0.086 (4) | 0.026 (2) | 0.038 (2) | 0.024 (2) | −0.001 (2) | 0.0030 (18) |
| O10 | 0.039 (2) | 0.042 (2) | 0.041 (2) | 0.0248 (19) | 0.0033 (18) | −0.0111 (18) |
| K1 | 0.0255 (5) | 0.0269 (5) | 0.0315 (6) | 0.0131 (4) | 0.0015 (4) | −0.0013 (5) |
| K2 | 0.0296 (5) | 0.0258 (5) | 0.0287 (5) | 0.0130 (4) | −0.0002 (5) | 0.0019 (4) |
| As1—O5 | 1.633 (4) | K1—O4ii | 3.129 (5) |
| As1—O6 | 1.691 (3) | K1—O5 | 2.784 (4) |
| As1—O7 | 1.703 (3) | K1—O7 | 3.450 (4) |
| As1—O4 | 1.708 (4) | K1—O8iv | 2.782 (6) |
| Cr1—O2 | 1.595 (4) | K1—O9 | 2.871 (5) |
| Cr1—O3 | 1.599 (4) | K1—O10v | 2.731 (7) |
| Cr1—O1 | 1.599 (4) | K2—O1vi | 2.845 (5) |
| Cr1—O4 | 1.840 (4) | K2—O2iii | 2.766 (4) |
| Cr2—O8 | 1.597 (4) | K2—O3vi | 3.042 (5) |
| Cr2—O9 | 1.599 (4) | K2—O4 | 3.076 (5) |
| Cr2—O10 | 1.601 (4) | K2—O6vii | 2.777 (5) |
| Cr2—O7 | 1.845 (3) | K2—O7 | 2.935 (4) |
| O5—H | 1.4683 | K2—O8iv | 3.127 (6) |
| K1—O1i | 2.724 (5) | K2—O9viii | 2.818 (4) |
| K1—O2ii | 2.830 (5) | K2—O10iv | 2.825 (5) |
| K1—O3iii | 2.731 (5) | ||
| O5···O6ii | 2.555 (5) | ||
| O5—As1—O6 | 108.9 (2) | O1—Cr1—O4 | 109.0 (2) |
| O5—As1—O7 | 112.40 (19) | O8—Cr2—O9 | 113.1 (3) |
| O6—As1—O7 | 110.30 (18) | O8—Cr2—O10 | 108.4 (2) |
| O5—As1—O4 | 116.5 (2) | O9—Cr2—O10 | 110.7 (3) |
| O6—As1—O4 | 108.8 (2) | O8—Cr2—O7 | 107.3 (2) |
| O7—As1—O4 | 99.66 (16) | O9—Cr2—O7 | 108.2 (2) |
| O2—Cr1—O3 | 112.8 (3) | O10—Cr2—O7 | 109.1 (2) |
| O2—Cr1—O1 | 111.6 (3) | As1—O4—Cr1 | 128.0 (2) |
| O3—Cr1—O1 | 107.9 (2) | As1—O5—H | 122.0 |
| O2—Cr1—O4 | 106.3 (2) | As1—O7—Cr2 | 128.4 (2) |
| O3—Cr1—O4 | 109.1 (2) |
| Symmetry codes: (i) x, y+1, z; (ii) −y, x−y, z−1/3; (iii) x+1, y+1, z; (iv) −y+1, x−y, z−1/3; (v) −y+1, x−y+1, z−1/3; (vi) −x+y+1, −x, z+1/3; (vii) x+1, y, z; (viii) x, y−1, z. |
Experimental details
| (I) | (II) | |
| Crystal data | ||
| Chemical formula | K2[HCr2AsO10] | K2[HCr2AsO10] |
| Mr | 418.13 | 418.13 |
| Crystal system, space group | Trigonal, P31 | Trigonal, P32 |
| Temperature (K) | 296 | 294 |
| a, c (Å) | 7.6931 (8), 14.623 (3) | 7.6963 (9), 14.6171 (11) |
| V (Å3) | 749.50 (19) | 749.82 (14) |
| Z | 3 | 3 |
| Radiation type | Mo Kα | Mo Kα |
| µ (mm−1) | 6.33 | 6.33 |
| Crystal size (mm) | 0.40 × 0.22 × 0.18 | 0.29 × 0.13 × 0.11 |
| Data collection | ||
| Diffractometer | Enraf-Nonius CAD-4 diffractometer | Enraf-Nonius CAD-4 diffractometer |
| Absorption correction | Analytical (de Meulenaer & Tompa, 1965) | – |
| Tmin, Tmax | 0.205, 0.320 | – |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 2063, 2051, 2040 | 1229, 1224, 1182 |
| Rint | 0.019 | 0.011 |
| (sin θ/λ)max (Å−1) | 0.703 | 0.649 |
| Refinement | ||
| R[F2 > 2σ(F2)], wR(F2), S | 0.050, 0.129, 1.29 | 0.021, 0.053, 1.08 |
| No. of reflections | 2051 | 1224 |
| No. of parameters | 137 | 137 |
| No. of restraints | 1 | 1 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 1.31, −1.08 | 0.46, −0.33 |
| Absolute structure | Flack (1983), with how many Friedel pairs | Flack (1983), with how many Friedel pairs |
| Absolute structure parameter | 0.17 (3) | 0.030 (16) |
Computer programs: CAD-4-PC Software (Enraf-Nonius, 1993), CAD-4-PC Software, TEXSAN (Molecular Structure Corporation, 1997), TEXSAN, SHELXL97 (Sheldrick, 1997), ATOMS (Dowty, 2003).

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural ChemistryThe full text of this article is available to subscribers to the journal.
- Information on subscribing
- Sample issue
- Purchase subscription
- Reduced-price subscriptions
- If you have already subscribed, you may need to register




The possibility that the potential barrier between the structure of K2HCr2AsO10 phase II (II—K2Cr2O7; Averbuch-Pouchot et al., 1978; hereinafter A—P) and that of paraelectric phase I is surmountable below the Curie temperature led to the prediction that phase II is ferroelectric (Abrahams, 2003). Crystals grown for associated physical property measurements were also used for an independent structural redetermination on two different crystals. Unsurprisingly, crystal 1 in space group P31 has a small enantiomorphic component, whereas crystal 2 in space group P32 is single-component (see the Refinement section for further discussion of this). \sch
The asymmetric unit of phase II contains one independent AsO3OH and two CrO4 tetrahedra, sharing O atoms to form individual HAsCr2O102− ions (Fig. 1a). The average Cr—O distance for the terminal O atoms in both crystals is 1.600 (4) Å (Table 1) and this does not differ significantly from the value of 1.612 (7) Å in II—K2Cr2O7 (Weakley et al., 2004), whereas the mean bridging Cr—O distance of 1.844 (4) Å in K2HCr2AsO10 very significantly exceeds the value of 1.785 (4) Å in II—K2Cr2O7.
Unlike the bond-length distribution in the CrO4 tetrahedra, the mean As—O distance of 1.632 (3) Å for the terminal O5 atom is significantly less than the mean As—O distance of 1.699 (9) Å for the two bridging atoms and the terminal O6 atom in the AsO3OH tetrahedron. The short inter-anionic contact O5···O6i [2.550 (11) and 2.555 (5) Å for crystals 1 and 2, respectively; symmetry code: (i) y-x, −x, z − 1/3 for crystal 1, −y, x-y, z − 1/2 for crystal 2] results from the formation of an O5···H···O6i bond between HAsCr2O102− ions. The arsenate distances given in Supplementary Table S1 show that the mean bridging and terminal As—O distances generally do not differ significantly, at 1.708 (20) and 1.682 (12) Å, and so the mean As—O distance may be taken as 1.69 (2) Å.
The bond-valence (BV) sums (Brown & Altermatt, 1985) for each atom in K2HCr2AsO10 are 5.03 (3) for As, 5.98 (4) for Cr1 and 5.96 (4) for Cr2, 1.327 (8) for K1 and 1.157 (6) for K2, 1.612 (14) for O5 and 1.400 (14) for O6, and 1.94–2.20 (3) for all other O atoms in crystal 2, with comparable values in crystal 1. The connectivity within the asymmetric unit is shown in Fig. 1(b), and the labelling for all atoms in the unit cell is given in Supplementary Fig. S1. The BV sum for As agrees with the expectated value, while those for Cr, K and O atoms other than O5 and O6 agree with the values observed in II—K2Cr2O7 (Weakley et al., 2004). The deficit of 0.99 in the joint valence of atoms O5 and O6 is clearly equivalent to an H atom which, while not located directly, results in the bond revealed in both crystals by the short O5···O6 distance. The H atoms form helices of radius ~0.60 Å about the 31 or 32 axes (Fig. 2), as they link HCr2AsO102− anions. H-atom location was not considered in the report by A—P; if taken as midway between O5 and O6i (Table 2), then the H atom in crystal 2 is at (0.0555, 0.0307, −0.0245). Since the BV sum for As—O6 is 1.23 (1), this bond is close to single, hence the H atom is most likely to be nearer O6.
Atomic coordinates (x',y',z') in the predicted supergroup P3121 or P3221 are derived from the coordinates of related pairs of atoms in P31 or P32 under the higher-symmetry constraint (Table 2). Maximum differences between the positions of independent atoms in phase II and those predicted in paraelectric phase I, following the necessary symmetry conversions, are 0.41 Å for H and 0.586 Å for O5 and O6, confirming the basis for the original prediction of ferroelectricity (Abrahams, 2003). [Atoms O9 and O10 in the designation of Averbuch-Puchot et al. (1978) are equivalent to atoms O5 and O6 in Table 2.] The predicted atomic arrangement in phase I is shown in Supplementary Figs. S2 and S3.
The formation of K2[CrO3AsO3OHCrO3] from its constituent ions (see Experimental) is possible only by fission of an O—Cr—O bond in the Cr2O72− anion and the presence of an AsO3OH2− ion. The two resulting As—O—Cr bonds form by elimination of O2− as H2O. The protonation of a terminal O atom on As results in chains of CrO3AsO3OHCrO32− ions linked through hydrogen bonds, as shown in Figs. 1(a) and 2. The potential barrier to these rearrangements appears to be surmounted only at aqueous reaction solution temperatures close to boiling.
Comparison of the three independent sets of atomic coordinates is made by plotting the ordered experimental quantiles Qexp = |ξi(1) - ξi(2)|/{σ2[ξi(1)] + σ2[ξi(2)]2}1/2 against the corresponding normal quantiles Qnorm, where ξi(1), ξi(2) are the ith parameters from the first and second independent determinations with the same setting, and σ[ξi(1)], σ[ξi(2)] are the corresponding standard uncertainties (s.u.s) of each parameter. In the absence of error, a linear array of unit slope and zero intercept results (Abrahams & Keve, 1971). The Qnorm magnitudes are conveniently calculated by the program NORMPA (Ross, 2003).
The 45 atomic-coordinate magnitudes determined with crystals 1 and 2 are compared in Fig. 3, the straight line giving the fit obtained by linear regression. The absence of outliers and the small departure of the slope from unity or linearity is indicative of minor systematic error in either coordinate set, but with joint s.u.s (j.s.u.s) that are underestimated by a factor of ~1.3. The corresponding normal probability comparison of each set with the A—P atomic coordinates (Supplementary Figs. S4 and S5) contrasts strongly with that in Fig. 3. Seven Qexp - Qnorm terms in each of the latter two cases depart strongly from linearity (Table 3), hence these may be rejected as outliers, since major departures from a normal distribution can be due only to major systematic error. The strong possibility of uncompensated enantiomorphous twinning giving rise to the strong parameter correlation in the least-squares refinement noted by A—P is one of several likely sources of error. The remaining 38 terms exhibit a slightly S-shaped distribution, with j.s.u.s underestimated by a factor of ~1.7 in the comparison of crystal 1 and A—P, and of ~2.0 for the comparison of crystal 2 and A—P. The correlation coefficient in all fits made by linear regression is 0.987–0.992. The experimental uncertainties should be corrected by factors not less than those derived from the magnitude of the slopes listed in Table 3; inclusion of outliers would clearly increase these factors.
The recommended phase-transition nomenclature (Tolédano et al., 1998, 2001) for phases I and II of K2HCr2AsO10, using the thermal values of Ylvisaker et al. (2001), is I|~590–540 K| P3221 (152) |Z = 3| non-ferroic | decomposes above ~590 K, II| 540–270 K| P32 (145) |Z = 3| ferroelectric| 2 variants, lower thermal limit not known.
Table 1. Selected distances (Å) in K2HCr2AsO10 phase II.
Table 2. Atomic coordinates of K2HCr2AsO10 in phase II and predicted coordinates in phase I, with component Δ(x) and total Δ(xyz) differences between the phases in Å.
Table 3. Linear-regression indicators for the K2HCr2AsO10 Qexp - Qnorm plots.