The crystal structure of the title complex, tetrakis[
![[mu]](/logos/entities/mu_rmgif.gif)
-6-amino-3-methyl-4-azahex-3-en-2-one oximato(1-)-
![[kappa]](/logos/entities/kappa_rmgif.gif) 4N
4N,
N',
N'':
O]tetracopper(II) tetraperchlorate 0.6-hydrate, [Cu
4(C
6H
12N
3O)
4](ClO
4)
4·0.6H
2O, shows the cation to be an oximate-bridged tetramer composed of four 6-amino-3-methyl-4-azahex-3-en-2-one oxime ligands and four copper(II) ions and to have crystallographically imposed
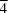
symmetry. Each Cu
II atom is four-coordinated by the three N atoms of one oxime ligand and by the O atom of another oxime ligand in a distorted square-planar geometry.
Supporting information
CCDC reference: 179257
The ligand 6-amino-3-methyl-4-azahex-3-en-2-one oxime was prepared according to
the method of Singh et al. (1977). Equimolar quantities of copper
perchlorate hexahydrate and the ligand were mixed in methanol and the solution
was heated under reflux for 2 h. The resulting solution was cooled to room
temperature and evaporated to dryness by rotatory evaporation. The crude
product was washed with acetonitrile and then recrystallized from methanol by
slow evaporation.
H atoms were placed geometrically and refined as riding, with Uiso(H) =
1.2Ueq(C, N or O). The water H atoms were not located. The largest
peak in the difference map is near Cu.
Data collection: CAD-4 Software (Enraf-Nonius, 1989); cell refinement: CAD-4 Software; data reduction: NRCVAX (Gabe et al., 1989); program(s) used to solve structure: SHELXS97 (Sheldrick, 1990) and NRCVAX; program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: NRCVAX; software used to prepare material for publication: SHELXL97.
Crystal data top
| [Cu4(C6H12N8O)4](ClO4)4·0.6H2O | Dx = 1.790 Mg m−3 |
| Mr = 1232.32 | Mo Kα radiation, λ = 0.71073 Å |
| Tetragonal, P42/n | Cell parameters from 25 reflections |
| a = 12.440 (3) Å | θ = 5.4–17.3° |
| c = 14.851 (7) Å | µ = 2.15 mm−1 |
| V = 2298.2 (12) Å3 | T = 293 K |
| Z = 2 | Square bipyramidal, black |
| F(000) = 1260 | 0.47 × 0.34 × 0.29 mm |
Data collection top
Nonius CAD-4
diffractometer | 1710 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.000 |
| Graphite monochromator | θmax = 30.0°, θmin = 2.1° |
| ω–2θ scans | h = 0→17 |
Absorption correction: empirical (using intensity measurements)
(North et al., 1968) | k = 0→17 |
| Tmin = 0.37, Tmax = 0.54 | l = 0→20 |
| 3349 measured reflections | 3 standard reflections every 60 min |
| 3349 independent reflections | intensity decay: 3% |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.066 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.183 | H-atom parameters constrained |
| S = 1.09 | w = 1/[σ2(Fo2) + (0.1P)2]
where P = (Fo2 + 2Fc2)/3 |
| 3349 reflections | (Δ/σ)max = 0.031 |
| 153 parameters | Δρmax = 1.31 e Å−3 |
| 0 restraints | Δρmin = −0.74 e Å−3 |
Crystal data top
| [Cu4(C6H12N8O)4](ClO4)4·0.6H2O | Z = 2 |
| Mr = 1232.32 | Mo Kα radiation |
| Tetragonal, P42/n | µ = 2.15 mm−1 |
| a = 12.440 (3) Å | T = 293 K |
| c = 14.851 (7) Å | 0.47 × 0.34 × 0.29 mm |
| V = 2298.2 (12) Å3 | |
Data collection top
Nonius CAD-4
diffractometer | 1710 reflections with I > 2σ(I) |
Absorption correction: empirical (using intensity measurements)
(North et al., 1968) | Rint = 0.000 |
| Tmin = 0.37, Tmax = 0.54 | 3 standard reflections every 60 min |
| 3349 measured reflections | intensity decay: 3% |
| 3349 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.066 | 0 restraints |
| wR(F2) = 0.183 | H-atom parameters constrained |
| S = 1.09 | Δρmax = 1.31 e Å−3 |
| 3349 reflections | Δρmin = −0.74 e Å−3 |
| 153 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | Occ. (<1) |
| Cu | 0.57965 (4) | 0.75901 (4) | 0.19143 (3) | 0.0455 (2) | |
| Cl | 0.36504 (11) | −0.05427 (12) | 0.27656 (10) | 0.0689 (4) | |
| O1 | 0.6149 (3) | 0.7823 (2) | 0.31724 (18) | 0.0464 (7) | |
| O2 | 0.4544 (3) | −0.0699 (4) | 0.2175 (3) | 0.0825 (12) | |
| O3 | 0.3256 (6) | −0.1522 (5) | 0.3059 (5) | 0.174 (3) | |
| O4 | 0.2841 (4) | 0.0031 (6) | 0.2328 (4) | 0.149 (2) | |
| O5 | 0.3975 (5) | 0.0029 (6) | 0.3523 (4) | 0.139 (2) | |
| OW | 0.7500 | 0.7500 | 0.6558 (15) | 0.221 (14) | 0.30 (3) |
| N1 | 0.4520 (3) | 0.6624 (3) | 0.2065 (3) | 0.0566 (10) | |
| H1A | 0.4722 | 0.5996 | 0.2314 | 0.068* | |
| H1B | 0.4031 | 0.6936 | 0.2428 | 0.068* | |
| N2 | 0.5295 (3) | 0.7720 (3) | 0.0683 (3) | 0.0552 (10) | |
| N3 | 0.6919 (3) | 0.8554 (3) | 0.1407 (2) | 0.0423 (8) | |
| C1 | 0.4056 (6) | 0.6438 (6) | 0.1174 (4) | 0.103 (3) | |
| H2A | 0.4331 | 0.5760 | 0.0947 | 0.123* | |
| H2B | 0.3284 | 0.6361 | 0.1241 | 0.123* | |
| C2 | 0.4252 (5) | 0.7224 (6) | 0.0538 (4) | 0.087 (2) | |
| H3A | 0.3696 | 0.7769 | 0.0572 | 0.104* | |
| H3B | 0.4230 | 0.6909 | −0.0059 | 0.104* | |
| C3 | 0.5803 (4) | 0.8361 (4) | 0.0179 (3) | 0.0574 (12) | |
| C4 | 0.5456 (6) | 0.8705 (6) | −0.0739 (4) | 0.103 (3) | |
| H4A | 0.4747 | 0.8435 | −0.0857 | 0.123* | |
| H4B | 0.5947 | 0.8426 | −0.1179 | 0.123* | |
| H4C | 0.5449 | 0.9476 | −0.0770 | 0.123* | |
| C5 | 0.7629 (5) | 0.9393 (5) | 0.0051 (4) | 0.0780 (18) | |
| H5A | 0.7460 | 1.0145 | 0.0068 | 0.094* | |
| H5B | 0.7630 | 0.9149 | −0.0563 | 0.094* | |
| H5C | 0.8327 | 0.9278 | 0.0310 | 0.094* | |
| C6 | 0.6812 (4) | 0.8782 (4) | 0.0573 (3) | 0.0487 (11) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Cu | 0.0509 (3) | 0.0544 (4) | 0.0312 (3) | −0.0061 (2) | −0.0043 (2) | 0.0055 (2) |
| Cl | 0.0638 (8) | 0.0758 (9) | 0.0669 (8) | 0.0017 (7) | 0.0049 (7) | −0.0011 (7) |
| O1 | 0.0584 (18) | 0.0494 (17) | 0.0315 (14) | 0.0025 (13) | −0.0040 (13) | 0.0049 (12) |
| O2 | 0.081 (3) | 0.090 (3) | 0.076 (3) | 0.015 (2) | 0.023 (2) | 0.014 (2) |
| O3 | 0.171 (7) | 0.103 (4) | 0.249 (9) | −0.026 (4) | 0.125 (6) | −0.014 (5) |
| O4 | 0.092 (4) | 0.259 (8) | 0.094 (4) | 0.063 (5) | −0.003 (3) | 0.014 (5) |
| O5 | 0.119 (4) | 0.217 (7) | 0.081 (3) | −0.030 (5) | 0.003 (3) | −0.062 (4) |
| OW | 0.25 (3) | 0.18 (2) | 0.23 (2) | −0.080 (17) | 0.000 | 0.000 |
| N1 | 0.063 (2) | 0.058 (2) | 0.049 (2) | −0.0108 (19) | −0.0078 (18) | 0.0078 (18) |
| N2 | 0.060 (2) | 0.067 (3) | 0.039 (2) | −0.011 (2) | −0.0141 (17) | 0.0044 (18) |
| N3 | 0.050 (2) | 0.0439 (18) | 0.0328 (17) | −0.0005 (15) | −0.0055 (15) | 0.0037 (14) |
| C1 | 0.103 (5) | 0.139 (6) | 0.066 (4) | −0.059 (5) | −0.027 (4) | 0.013 (4) |
| C2 | 0.075 (4) | 0.128 (6) | 0.058 (3) | −0.041 (4) | −0.021 (3) | 0.017 (4) |
| C3 | 0.077 (3) | 0.060 (3) | 0.035 (2) | −0.007 (2) | −0.012 (2) | 0.005 (2) |
| C4 | 0.123 (6) | 0.139 (6) | 0.046 (3) | −0.038 (5) | −0.034 (4) | 0.032 (4) |
| C5 | 0.096 (4) | 0.097 (4) | 0.041 (3) | −0.031 (3) | −0.003 (3) | 0.019 (3) |
| C6 | 0.063 (3) | 0.052 (3) | 0.032 (2) | −0.005 (2) | −0.0030 (19) | 0.0029 (17) |
Geometric parameters (Å, º) top
| Cu—N2 | 1.939 (4) | N3—O1ii | 1.339 (4) |
| Cu—O1 | 1.941 (3) | C1—C2 | 1.382 (8) |
| Cu—N3 | 1.989 (3) | C1—H2A | 0.9700 |
| Cu—N1 | 2.004 (4) | C1—H2B | 0.9700 |
| Cl—O3 | 1.383 (6) | C2—H3A | 0.9700 |
| Cl—O5 | 1.391 (5) | C2—H3B | 0.9700 |
| Cl—O4 | 1.395 (6) | C3—C6 | 1.480 (6) |
| Cl—O2 | 1.429 (4) | C3—C4 | 1.492 (6) |
| O1—N3i | 1.339 (4) | C4—H4A | 0.9600 |
| N1—C1 | 1.462 (7) | C4—H4B | 0.9600 |
| N1—H1A | 0.9000 | C4—H4C | 0.9600 |
| N1—H1B | 0.9000 | C5—C6 | 1.487 (7) |
| N2—C3 | 1.264 (6) | C5—H5A | 0.9600 |
| N2—C2 | 1.452 (6) | C5—H5B | 0.9600 |
| N3—C6 | 1.278 (5) | C5—H5C | 0.9600 |
| | | |
| N2—Cu—O1 | 165.50 (16) | C2—C1—H2B | 108.3 |
| N2—Cu—N3 | 79.54 (15) | N1—C1—H2B | 108.3 |
| O1—Cu—N3 | 96.66 (15) | H2A—C1—H2B | 107.4 |
| N2—Cu—N1 | 84.26 (16) | C1—C2—N2 | 110.9 (5) |
| O1—Cu—N1 | 99.28 (16) | C1—C2—H3A | 109.5 |
| N3—Cu—N1 | 163.78 (15) | N2—C2—H3A | 109.4 |
| O3—Cl—O5 | 107.4 (5) | C1—C2—H3B | 109.4 |
| O3—Cl—O4 | 110.0 (4) | N2—C2—H3B | 109.5 |
| O5—Cl—O4 | 108.9 (4) | H3A—C2—H3B | 108.0 |
| O3—Cl—O2 | 110.4 (3) | N2—C3—C6 | 114.4 (4) |
| O5—Cl—O2 | 109.9 (3) | N2—C3—C4 | 125.3 (5) |
| O4—Cl—O2 | 110.2 (3) | C6—C3—C4 | 120.3 (5) |
| N3i—O1—Cu | 112.7 (2) | C3—C4—H4A | 109.5 |
| C1—N1—Cu | 107.9 (3) | C3—C4—H4B | 109.5 |
| C1—N1—H1A | 110.1 | H4A—C4—H4B | 109.5 |
| Cu—N1—H1A | 110.1 | C3—C4—H4C | 109.5 |
| C1—N1—H1B | 110.2 | H4A—C4—H4C | 109.5 |
| Cu—N1—H1B | 110.1 | H4B—C4—H4C | 109.5 |
| H1A—N1—H1B | 108.4 | C6—C5—H5A | 109.5 |
| C3—N2—C2 | 128.8 (4) | C6—C5—H5B | 109.5 |
| C3—N2—Cu | 116.8 (3) | H5A—C5—H5B | 109.5 |
| C2—N2—Cu | 113.1 (3) | C6—C5—H5C | 109.5 |
| C6—N3—O1ii | 118.6 (3) | H5A—C5—H5C | 109.5 |
| C6—N3—Cu | 115.3 (3) | H5B—C5—H5C | 109.5 |
| O1ii—N3—Cu | 125.4 (2) | N3—C6—C3 | 113.1 (4) |
| C2—C1—N1 | 115.9 (5) | N3—C6—C5 | 123.2 (4) |
| C2—C1—H2A | 108.3 | C3—C6—C5 | 123.7 (4) |
| N1—C1—H2A | 108.3 | | |
| Symmetry codes: (i) −y+3/2, x, −z+1/2; (ii) y, −x+3/2, −z+1/2. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···O2iii | 0.90 | 2.31 | 3.181 (6) | 164 |
| N1—H1B···O3iv | 0.90 | 2.34 | 3.158 (8) | 151 |
| OW—(H)···O4v | | | 3.305 (10) | |
| Symmetry codes: (iii) −y+1/2, x, −z+1/2; (iv) x, y+1, z; (v) −y+1, x+1/2, z+1/2. |
Experimental details
| Crystal data |
| Chemical formula | [Cu4(C6H12N8O)4](ClO4)4·0.6H2O |
| Mr | 1232.32 |
| Crystal system, space group | Tetragonal, P42/n |
| Temperature (K) | 293 |
| a, c (Å) | 12.440 (3), 14.851 (7) |
| V (Å3) | 2298.2 (12) |
| Z | 2 |
| Radiation type | Mo Kα |
| µ (mm−1) | 2.15 |
| Crystal size (mm) | 0.47 × 0.34 × 0.29 |
| |
| Data collection |
| Diffractometer | Nonius CAD-4
diffractometer |
| Absorption correction | Empirical (using intensity measurements)
(North et al., 1968) |
| Tmin, Tmax | 0.37, 0.54 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 3349, 3349, 1710 |
| Rint | 0.000 |
| (sin θ/λ)max (Å−1) | 0.703 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.066, 0.183, 1.09 |
| No. of reflections | 3349 |
| No. of parameters | 153 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 1.31, −0.74 |
Selected geometric parameters (Å, º) top| Cu—N2 | 1.939 (4) | Cu—N3 | 1.989 (3) |
| Cu—O1 | 1.941 (3) | Cu—N1 | 2.004 (4) |
| | | |
| N2—Cu—O1 | 165.50 (16) | N2—Cu—N1 | 84.26 (16) |
| N2—Cu—N3 | 79.54 (15) | O1—Cu—N1 | 99.28 (16) |
| O1—Cu—N3 | 96.66 (15) | N3—Cu—N1 | 163.78 (15) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···O2i | 0.90 | 2.305 | 3.181 (6) | 164.2 |
| N1—H1B···O3ii | 0.90 | 2.343 | 3.158 (8) | 150.5 |
| OW—(H)···O4iii | . | . | 3.305 (10) | . |
| Symmetry codes: (i) −y+1/2, x, −z+1/2; (ii) x, y+1, z; (iii) −y+1, x+1/2, z+1/2. |


 journal menu
journal menu metal-organic compounds
metal-organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2001/12/00/ta1319/ta1319fig1thm.gif)




Studies on multinuclear copper(II) complexes have been focused on the magneto-structural relationships and characterization of the active site in multicopper proteins (Alagi et al., 1997). Linear, di- and trinuclear copper(II) complexes with oximate groups (C = N—O-) have been widely studied (Cervera et al., 1997; Dominguezvera et al., 1997; Luneau et al., 1989), but tetranuclear copper(II) complexes occur less frequently. In the present study, the isolation and X-ray structure of the tetranuclear copper(II) complex tetrakis[µ-6-amino-3-methyl-4-azahex-3-en-2-one oximato(1-)-κ4N,N',N'':O]- tetracopper(II) tetraperchlorate 0.6-hydrate, (I), is reported.
Each Cu ion in the structure is coordinated by three N atoms from one 6-amino-3-methyl-4-aza-hex-3-en-2-one oximate ligand (L-) and by the oxime O atom from a second ligand (L-) in a distorted square-planar arrangement. The O1, N1, N2 and N3 donor atoms are planar within 0.124 (2) Å, and the Cu atom deviates by 0.129 (2) Å from this plane. Four of these CuL subunits are linked into a discrete tetrameric cation which has 4 crystallographic symmetry (Fig. 1) and which contains a 12-membered heterocyclic ring of composition (Cu,O1,N3)4. The unit cell contains two of these tetramers.
Unlike the structures of square tetranuclear metal complexes in which the four metal ions lie in a plane (Chaudhuri et al., 1993; Maekawa et al., 1999), the central tetranuclear copper(II) core of the title complex has an `open-butterfly' configuration with the four copper(II) ions located at the corners of a flattened tetrahedron and is much different from rhombic structures (Raper, 1997; Castro et al., 1995; Gomez-Garcia et al., 1992; Sletten et al., 1990; Tandon et al., 1991). The degree of distortion of the tetrahedron is indicated by the two Cu···Cu distances of 4.244 (1) Å perpendicular to the c axis and the four other Cu···Cu distances of 3.469 (1) Å within the tetramer. The oxime group adopts an out-of-plane coordination mode as a diatomic (µ-1,2)-bridging ligand between copper(II) ions (Ruiz et al., 1998). The orientations of the oxime ligands in the tetramer are alternately up and down.
The Cu/N1/N2/C1/C2 and Cu/N1/N2/C1/C2 five-membered rings are in the skew form and are planar within 0.158 (5) and 0.051 (3) Å, respectively. The Cu— Nbond lengths, ranging from 1.939 (4) to 2.004 (4) Å, are considered normal covalent bonds. The order of the Cu—N distances, amine [2.004 (4) Å] > oxime [1.989 (3) Å] > imine [1.941 (3) Å], is the same as that observed for other oximato-bridged CuII complexes (Nasakkala et al., 1981; Butcher et al., 1979). The Cu—O(bridging oxime) distance [1.941 (3) Å] is slightly longer than that found in these oximate-bridged CuII complexes because of the steric effect of the bulky folded conformation (Nasakkala et al., 1981; Butcher et al., 1979). Owing to the weak metal–ligand interactions in the title complex, the N—O distance [1.339 (4) Å] is longer than that found in these complexes with strongly coordinated bridging-oxime groups [1.311 (3) Å; Raston et al., 1978; Butcher et al., 1979]. The bond angles of the oxime and imine N atoms are approximately 120°, in keeping with the expected sp2-hybridization stabilized by π-bonding. The N3i—O1—Cu [112.7 (2)°; symmetry code: (i) 3/2 - y, x, 1/2 - z] angle indicates a variation from sp2 hybridization for the oxime O atom, resulting from coordination to another Cu atom. The N—O distance does show some decrease in going from the free ligand (1.375 Å) to complexes with weak interactions [1.339 (4) Å, this study] to complexes with strong interactions [1.311 (3) Å; Raston et al., 1978; Butcher et al., 1979].
There appears to be a water molecule on a partially occupied site [occupancy = 0.30 (3)] on a twofold axis. This is external to the tetramer and is loosely held by hydrogen bonding to the perchlorate ion (Table 2). There are two equivalent sites on the axis that are 2.80 (3) Å apart. Other hydrogen bonds are detailed in Table 2.