
Two different forms of
meso-3,3'-[2,2-dimethylpropane-1,3-diylbis(azanediyl)]dibutan-2-one dioxime, commonly called
meso-hexamethyl propylene amine oxime (HMPAO), C
13H
28N
4O
2, designated
![[alpha]](/logos/entities/alpha_rmgif.gif)
and
![[beta]](/logos/entities/beta_rmgif.gif)
, were isolated by fractional crystallization and their crystal structures were determined by powder X-ray diffraction using the direct-space method with the parallel tempering algorithm. The
form was first crystallized from acetonitrile solution, while the
form was obtained by recrystallization of the
phase from diethyl ether. The
form crystallizes in the triclinic system (space group
P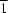
), with one molecule in the asymmetric unit, while the crystal of the
form is monoclinic (space group
P2
1/
n), with one molecule in the asymmetric unit. In both phases, the molecules have similar conformations and
RS/
EE geometric isomerism. The crystal packing of the two phases is dominated by intermolecular hydrogen-bonding interactions between the two O-H oxime groups of an individual molecule and the amine N atoms of two different adjacent molecules, which lead to segregation of extended poly(
meso-HMPAO) one-dimensional chains along the
c direction. The structures of the two phases are primarily different due to the different orientations of the molecules in the chains.
Supporting information
CCDC references: 796083; 796084
All reactions and manipulations were carried out under an inert atmosphere using
a two-fold vacuum line and Schlenk techniques. 1H and 13C{1H} NMR
spectra were recorded on a Bruker Biospin 400 spectrometer in CDCl3. IR
spectra were recorded on a Jasco FTIR 300E instrument. Microanalysis was
performed using a EURO EA analyser. X-ray powder diffraction patterns were
obtained on a Stoe STADI P diffractometer with monochromatic Cu Kα1
radiation (λ = 1.5406 Å) selected using an incident-beam curved-crystal
germanium Ge(111) monochromator, using the Stoe transmission geometry
(horizontal set-up) with a linear position-sensitive detector (PSD). Melting
points were determined by differential thermal analysis measurements on a
Netzsch DTA 404 EP instrument. The diastereomeric mixture of HMPAO was
prepared and separated by fractional crystallization according to the
published method (Banerjee et al., 1999). Form (Iα) of HMPAO
was first
crystallized from acetonitrile solution at room temperature, while form (Iβ)
was obtained by recrystallization of (Iα) from Et2O at room temperature.
The purities of the two meso-HMPAO phases were confirmed by
multinuclear NMR and IR spectroscopic techniques and microanalysis. Many
attempts were made to grow high-quality crystals of meso-HMPAO
suitable for single X-ray diffraction study, but without success.
The powder of (Iα) or (Iβ) was ground, placed between two foils of Mylar, and
fixed in the sample holder with a mask of suitable internal diameter (0.7 mm).
The pattern of the α form was scanned over the angular range 3–80° (2θ)
with a step width of the PSD of 0.1° (2θ) and a counting time of 60 s per
step, while the pattern of the β form was scanned over the angular range
5–70° (2θ) with a step width of the PSD of 0.5° (2θ) and a counting time
of 420 s per step. Pattern indexing was performed using the DICVOL4.0
program (Boultif & Louër, 2004) with default options. Confidence
factors were
M(20) = 34.1 and F(20) = 96.4 for the α form, and M(20)
= 32.4 and F(20) = 85.4 for the β form. The space groups were obtained
using the program CHECK-CELL interfaced by WINPLOTR (Roisnel &
Rodriguez-Carvajal, 2001). The unreduced triclinic cell for (Iα) was
used to
make the c axis the common axis for the molecular chains in the two
phases.
Direct methods were initially employed to determine the crystal structures
using the program EXPO2004 (Altomare et al., 1999), but
they
were not successful. Starting models for the two forms were obtained using the
direct-space method with the parallel tempering algorithm in the program
FOX (Favre-Nicolin & Černý, 2002). One molecule of
meso-HMPAO
for both phases was introduced randomly with the possibility to translate, to
rotate around its centre of mass and to modify its ten torsion angles. The
degree of freedom for the molecular replacement for the two forms is 16. In
order to accelerate the process during the parallel tempering calculation, the
powder patterns were truncated to 36° (Cu Kα1) and the H atoms were
not introduced. This method yielded a suitable model with agreement factors of
Rp = 0.0776 for (Iα) and Rp = 0.0459 for (Iβ).
The two models thus obtained were used as starting points for Rietveld
refinements in the program GSAS (Larson & Von Dreele, 2004),
interfaced
by EXPGUI (Toby, 2001). The coordinates of the 19 non-H atoms
were
refined with soft constraints on bond lengths. The profile function used was a
pseudo-Voigt function convoluted with an axial divergence asymmetry function
(Finger et al., 1994), and with S/L and
D/L
both fixed at 0.0225.
An isotropic displacement parameter was introduced and refined for each type of
atom. Intensities were corrected for absorption effects with an m.d.
[Define?] value of 0.1600 for (Iα) and 0.0434 for (Iβ). Before the
final refinement, H atoms of the CH, CH2 and CH3 groups were introduced
from geometric arguments and refined with constraints to the riding atoms
(0.99 Å for CH, 0.98 Å for CH2 and 0.97 Å for CH3). The H atoms of
the hydroxyl and amine groups were localized by difference Fourier syntheses
and refined with constraints on bond lengths (0.82 Å for OH and 0.87 Å for
NH) and angles. The background was refined using a shifted Chebyshev
polynomial with 15 coefficients, whereas the preferred orientation was
modelled using the generalized spherical-harmonics description (Von Dreele,
1997).
Final agreement factors are provided in the experimental tables. Fig. 4 shows
the experimental X-ray patterns, together with the calculated patterns and
difference curves from the final Rietveld refinements for both forms.
For both compounds, data collection: WINXPOW (Stoe & Cie, 1999); cell refinement: GSAS (Larson & Von Dreele, 2004); data reduction: WINXPOW (Stoe & Cie, 1999); program(s) used to solve structure: FOX (Favre-Nicolin & Černý, 2002); program(s) used to refine structure: GSAS (Larson & Von Dreele, 2004); molecular graphics: ORTEP-3 (Farrugia, 1997); software used to prepare material for publication: publCIF (Westrip, 2010).
(Ialpha)
meso-3,3'-[2,2-dimethylpropane-1,3-diylbis(azanediyl)]dibutan-2-one
dioxime
top
Crystal data top
| C13H28N4O2 | Z = 2 |
| Mr = 272.39 | F(000) = 300 |
| Triclinic, P1 | Dx = 1.108 Mg m−3 |
| Hall symbol: -p 1 | Cu Kα1 radiation, λ = 1.5406 Å |
| a = 15.3159 (3) Å | µ = 0.61 mm−1 |
| b = 8.74371 (12) Å | T = 298 K |
| c = 6.21770 (9) Å | Particle morphology: needle (visual estimate) |
| α = 81.1652 (10)° | white |
| β = 93.4835 (10)° | flat sheet, 7 × 7 mm |
| γ = 96.4282 (8)° | Specimen preparation: Prepared at 298 K and 101.3 kPa |
| V = 816.80 (2) Å3 | |
Data collection top
Stoe STADI P
diffractometer | Data collection mode: transmission |
| Radiation source: sealed X-ray tube, C-Tech | Scan method: step |
| Ge 111 monochromator | 2θmin = 2.992°, 2θmax = 79.982°, 2θstep = 0.01° |
| Specimen mounting: drifted powder between two Mylar foils | |
Refinement top
| Least-squares matrix: full | Profile function: CW Profile function number 4 with 27 terms
Pseudovoigt profile coefficients as parameterized in Thompson et al.
(1987).
Asymmetry correction of (Finger et al., 1994).
Microstrain broadening (Stephens, 1999).
#1(GU) = 0.000 #2(GV) = 0.000 #3(GW) = 10.581
#4(GP) = 0.000 #5(LX) = 3.335 #6(ptec) = 0.00
#7(trns) = 0.00 #8(shft) = 0.0000 #9(sfec) = 0.00
#10(S/L) = 0.0225 #11(H/L) = 0.0225 #12(eta) = 0.5000
Peak tails are ignored where the intensity is below 0.0010 times the peak.
Aniso. broadening axis 0.0 0.0 1.0
Stephens, P. W. (1999). J. Appl. Cryst. 32, 281–289.
Thompson, P., Cox, D. E. & Hastings, J. B. (1987). J. Appl.
Cryst.
20, 79–83. |
| Rp = 0.017 | 224 parameters |
| Rwp = 0.023 | 104 restraints |
| Rexp = 0.019 | H-atom parameters constrained |
| R(F2) = 0.01476 | (Δ/σ)max = 0.02 |
| χ2 = 1.416 | Background function: GSAS background function number 1 with 20 terms.
Shifted Chebyshev function of 1st kind
1: 2095.63 2: -2093.54 3: 1003.99 4: -224.356
5: -35.0151 6: 46.3120 7: 9.05002 8: -15.5584
9: -4.55608 10: 16.8935 11: -8.33517 12: 3.12503
13: -0.184876 14: -14.2318 15: 26.9383 16: -17.5033
17: 5.55156 18: -2.00997 19: 10.5513 20: -2.64504 |
| 7700 data points | |
Crystal data top
| C13H28N4O2 | γ = 96.4282 (8)° |
| Mr = 272.39 | V = 816.80 (2) Å3 |
| Triclinic, P1 | Z = 2 |
| a = 15.3159 (3) Å | Cu Kα1 radiation, λ = 1.5406 Å |
| b = 8.74371 (12) Å | µ = 0.61 mm−1 |
| c = 6.21770 (9) Å | T = 298 K |
| α = 81.1652 (10)° | flat sheet, 7 × 7 mm |
| β = 93.4835 (10)° | |
Data collection top
Stoe STADI P
diffractometer | Scan method: step |
| Specimen mounting: drifted powder between two Mylar foils | 2θmin = 2.992°, 2θmax = 79.982°, 2θstep = 0.01° |
| Data collection mode: transmission | |
Refinement top
| Rp = 0.017 | 7700 data points |
| Rwp = 0.023 | 224 parameters |
| Rexp = 0.019 | 104 restraints |
| R(F2) = 0.01476 | H-atom parameters constrained |
| χ2 = 1.416 | |
Special details top
Experimental. The sample was ground lightly in a mortar, loaded between two Mylar foils and
fixed in the sample holder with a mask of 7.0 mm internal diameter. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| C1 | 0.8535 (3) | 0.2572 (5) | 0.5458 (7) | 0.0384 (14)* | |
| C2 | 0.8898 (2) | 0.4043 (4) | 0.6396 (5) | 0.0384 (14)* | |
| C3 | 0.9297 (2) | 0.1916 (4) | 0.4536 (6) | 0.0384 (14)* | |
| C4 | 0.7864 (2) | 0.2921 (4) | 0.3527 (6) | 0.0384 (14)* | |
| C5 | 0.8146 (2) | 0.1333 (4) | 0.7298 (6) | 0.0384 (14)* | |
| N1 | 0.7022 (3) | 0.3372 (5) | 0.4180 (8) | 0.040 (2)* | |
| N2 | 0.7779 (3) | −0.0133 (5) | 0.6474 (7) | 0.040 (2)* | |
| C6 | 0.6527 (3) | 0.4088 (5) | 0.2256 (7) | 0.0384 (14)* | |
| C7 | 0.7319 (3) | −0.1032 (5) | 0.8360 (7) | 0.0384 (14)* | |
| C8 | 0.5772 (2) | 0.4852 (4) | 0.3012 (5) | 0.0384 (14)* | |
| C9 | 0.6775 (2) | −0.2449 (4) | 0.7572 (5) | 0.0384 (14)* | |
| C10 | 0.6267 (6) | 0.2786 (7) | 0.0933 (10) | 0.0384 (14)* | |
| C11 | 0.7902 (4) | −0.1690 (9) | 1.0215 (10) | 0.0384 (14)* | |
| C12 | 0.5702 (2) | 0.1332 (4) | 0.2004 (6) | 0.0384 (14)* | |
| C13 | 0.8665 (2) | −0.2550 (4) | 0.9718 (5) | 0.0384 (14)* | |
| N3 | 0.6410 (5) | 0.3115 (9) | −0.1105 (10) | 0.040 (2)* | |
| N4 | 0.7815 (5) | −0.1252 (9) | 1.2052 (10) | 0.040 (2)* | |
| O1 | 0.6002 (5) | 0.1967 (6) | −0.2299 (8) | 0.048 (2)* | |
| O2 | 0.8376 (5) | −0.1993 (7) | 1.3625 (8) | 0.048 (2)* | |
| H2A | 0.88814 | 0.49396 | 0.52684 | 0.05* | |
| H2B | 0.85425 | 0.41718 | 0.75794 | 0.05* | |
| H2C | 0.94988 | 0.39484 | 0.69383 | 0.05* | |
| H3A | 0.95355 | 0.26587 | 0.33346 | 0.05* | |
| H3B | 0.9755 | 0.17345 | 0.56739 | 0.05* | |
| H3C | 0.90859 | 0.09428 | 0.40168 | 0.05* | |
| H4A | 0.77375 | 0.19927 | 0.28132 | 0.05* | |
| H4B | 0.81331 | 0.37719 | 0.24823 | 0.05* | |
| H5A | 0.86107 | 0.10959 | 0.84165 | 0.05* | |
| H5B | 0.76775 | 0.17592 | 0.79519 | 0.05* | |
| H6A | 0.69258 | 0.49015 | 0.14125 | 0.05* | |
| H7A | 0.69202 | −0.03753 | 0.89032 | 0.05* | |
| H8A | 0.54135 | 0.40887 | 0.39801 | 0.05* | |
| H8B | 0.60102 | 0.57022 | 0.37816 | 0.05* | |
| H8C | 0.54136 | 0.52499 | 0.17544 | 0.05* | |
| H9A | 0.71659 | −0.31722 | 0.72425 | 0.05* | |
| H9B | 0.63901 | −0.29623 | 0.87054 | 0.05* | |
| H9C | 0.64248 | −0.21037 | 0.62731 | 0.05* | |
| H12A | 0.60318 | 0.04332 | 0.21278 | 0.05* | |
| H12B | 0.55505 | 0.14876 | 0.34422 | 0.05* | |
| H12C | 0.51667 | 0.11546 | 0.11178 | 0.05* | |
| H13A | 0.90952 | −0.18216 | 0.89097 | 0.05* | |
| H13B | 0.84452 | −0.33576 | 0.88576 | 0.05* | |
| H13C | 0.89393 | −0.30161 | 1.10752 | 0.05* | |
| H2O | 0.84791 | −0.14763 | 1.46207 | 0.05* | |
| H2N | 0.73981 | 0.00769 | 0.54043 | 0.05* | |
| H1O | 0.59915 | 0.23172 | −0.36018 | 0.05* | |
| H1N | 0.71277 | 0.40271 | 0.51053 | 0.05* | |
Geometric parameters (Å, º) top
| C1—C2 | 1.528 (4) | C6—H6A | 0.991 |
| C1—C3 | 1.535 (5) | C7—C9 | 1.541 (4) |
| C1—C4 | 1.548 (4) | C7—C11 | 1.498 (5) |
| C1—C5 | 1.548 (5) | C7—H7A | 0.9910 |
| C2—C1 | 1.528 (4) | C8—H8A | 0.9715 |
| C2—H2A | 0.9696 | C8—H8B | 0.9708 |
| C2—H2B | 0.9683 | C8—H8C | 0.9697 |
| C2—H2C | 0.9687 | C9—H9A | 0.9696 |
| C3—H3A | 0.9717 | C9—H9B | 0.9696 |
| C3—H3B | 0.9732 | C9—H9C | 0.9695 |
| C3—H3C | 0.9711 | C10—C12 | 1.540 (5) |
| C4—N1 | 1.487 (5) | C10—N3 | 1.282 (5) |
| C4—H4A | 0.979 | C11—C13 | 1.529 (5) |
| C4—H4B | 0.980 | C11—N4 | 1.278 (5) |
| C5—H5A | 0.981 | C12—H12A | 0.9720 |
| C5—H5B | 0.979 | C12—H12B | 0.9701 |
| N1—C4 | 1.487 (5) | C12—H12C | 0.9705 |
| N1—C6 | 1.471 (5) | C13—H13A | 0.9699 |
| N1—H1N | 0.869 | C13—H13B | 0.9689 |
| N2—C5 | 1.492 (4) | C13—H13C | 0.9714 |
| N2—C7 | 1.477 (5) | N3—O1 | 1.407 (5) |
| N2—H2N | 0.869 | N4—O2 | 1.394 (5) |
| C6—C8 | 1.530 (4) | O1—H1O | 0.820 |
| C6—C10 | 1.510 (5) | O2—H2O | 0.819 |
| | | |
| C2—C1—C3 | 108.4 (3) | C10—C6—H6A | 109.2 |
| C2—C1—C4 | 111.9 (3) | N2—C7—C9 | 107.6 (3) |
| C2—C1—C5 | 109.6 (3) | N2—C7—C11 | 115.5 (5) |
| C3—C1—C4 | 106.3 (3) | N2—C7—H7A | 109.4 |
| C3—C1—C5 | 108.0 (3) | C9—C7—C11 | 105.5 (4) |
| C4—C1—C5 | 112.5 (3) | C9—C7—H7A | 109.40 |
| C1—C2—H2A | 109.54 | C11—C7—H7A | 109.10 |
| C1—C2—H2B | 109.42 | C6—C8—H8A | 109.58 |
| C1—C2—H2C | 109.32 | C6—C8—H8B | 109.42 |
| H2A—C2—H2B | 109.40 | C6—C8—H8C | 109.35 |
| H2A—C2—H2C | 109.68 | H8A—C8—H8B | 109.39 |
| H2B—C2—H2C | 109.47 | H8A—C8—H8C | 109.55 |
| C1—C3—H3A | 109.36 | H8B—C8—H8C | 109.54 |
| C1—C3—H3B | 109.41 | C7—C9—H9A | 109.67 |
| C1—C3—H3C | 109.63 | C7—C9—H9B | 109.43 |
| H3A—C3—H3B | 109.48 | C7—C9—H9C | 109.46 |
| H3A—C3—H3C | 109.39 | H9A—C9—H9B | 109.39 |
| H3B—C3—H3C | 109.56 | H9A—C9—H9C | 109.44 |
| C1—C4—N1 | 113.7 (4) | H9B—C9—H9C | 109.44 |
| C1—C4—H4A | 108.61 | C6—C10—C12 | 118.1 (4) |
| C1—C4—H4B | 108.30 | C6—C10—N3 | 115.5 (5) |
| N1—C4—H4A | 108.68 | C12—C10—N3 | 125.2 (6) |
| N1—C4—H4B | 108.7 | C7—C11—C13 | 118.7 (4) |
| H4A—C4—H4B | 108.7 | C7—C11—N4 | 116.2 (5) |
| C1—C5—N2 | 112.0 (4) | C13—C11—N4 | 123.9 (6) |
| C1—C5—H5A | 108.91 | C10—C12—H12A | 109.40 |
| C1—C5—H5B | 108.83 | C10—C12—H12B | 109.51 |
| N2—C5—H5A | 108.95 | C10—C12—H12C | 109.70 |
| N2—C5—H5B | 109.18 | H12A—C12—H12B | 109.58 |
| H5A—C5—H5B | 108.9 | H12A—C12—H12C | 109.34 |
| C4—N1—C6 | 110.1 (4) | H12B—C12—H12C | 109.35 |
| C4—N1—H1N | 109.7 | C11—C13—H13A | 109.40 |
| C6—N1—H1N | 109.5 | C11—C13—H13B | 109.50 |
| C5—N2—C7 | 103.2 (4) | C11—C13—H13C | 109.48 |
| C5—N2—H2N | 109.7 | H13A—C13—H13B | 109.46 |
| C7—N2—H2N | 109.3 | H13A—C13—H13C | 109.45 |
| N1—C6—C8 | 108.9 (3) | H13B—C13—H13C | 109.54 |
| N1—C6—C10 | 104.8 (5) | C10—N3—O1 | 112.3 (7) |
| N1—C6—H6A | 108.7 | C11—N4—O2 | 109.7 (6) |
| C8—C6—C10 | 116.1 (5) | N3—O1—H1O | 109.3 |
| C8—C6—H6A | 108.8 | N4—O2—H2O | 109.5 |
| | | |
| C6—N1—C4—C1 | 164.5 (3) | C2—C1—C5—N2 | 179.9 (3) |
| C4—N1—C6—C8 | −167.9 (3) | C5—C1—C4—N1 | 54.4 (4) |
| C4—N1—C6—C10 | 67.2 (5) | C3—C1—C4—N1 | 172.3 (3) |
| C5—N2—C7—C11 | −72.3 (5) | C2—C1—C4—N1 | −69.5 (4) |
| C7—N2—C5—C1 | −170.9 (3) | N1—C6—C10—C12 | 59.1 (7) |
| C5—N2—C7—C9 | 170.2 (3) | C8—C6—C10—C12 | −61.1 (7) |
| O1—N3—C10—C12 | −1.2 (11) | C8—C6—C10—N3 | 107.1 (8) |
| O1—N3—C10—C6 | −168.5 (6) | N1—C6—C10—N3 | −132.6 (7) |
| O2—N4—C11—C13 | −14.5 (10) | N2—C7—C11—C13 | −47.4 (7) |
| O2—N4—C11—C7 | 178.1 (6) | C9—C7—C11—N4 | −120.6 (7) |
| C3—C1—C5—N2 | −62.1 (4) | C9—C7—C11—C13 | 71.3 (6) |
| C4—C1—C5—N2 | 54.8 (4) | N2—C7—C11—N4 | 120.7 (6) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1O···N1i | 0.82 | 2.19 | 2.811 (8) | 133 |
| O2—H2O···N2ii | 0.82 | 2.17 | 2.834 (7) | 138 |
| Symmetry codes: (i) x, y, z−1; (ii) x, y, z+1. |
(Ibeta)
meso-3,3'-[2,2-dimethylpropane-1,3-diylbis(azanediyl)]dibutan-2-one
dioxime
top
Crystal data top
| C13H28N4O2 | F(000) = 600 |
| Mr = 272.39 | Dx = 1.116 Mg m−3 |
| Monoclinic, P21/n | Cu Kα1 radiation, λ = 1.5406 Å |
| Hall symbol: -p 2yn | µ = 0.61 mm−1 |
| a = 16.4453 (4) Å | T = 298 K |
| b = 15.9587 (3) Å | Particle morphology: fine powder (visual estimate) |
| c = 6.20073 (11) Å | white |
| β = 94.7097 (13)° | flat sheet, 7 × 7 mm |
| V = 1621.86 (7) Å3 | Specimen preparation: Prepared at 298 K and 101.3 kPa |
| Z = 4 | |
Data collection top
Stoe STADI P
diffractometer | Data collection mode: transmission |
| Radiation source: sealed X-ray tube, C-Tech | Scan method: step |
| Ge 111 monochromator | 2θmin = 5.970°, 2θmax = 69.960°, 2θstep = 0.01° |
| Specimen mounting: drifted powder between two Mylar foils | |
Refinement top
| Least-squares matrix: full | Profile function: CW Profile function number 4 with 21 terms
Pseudovoigt profile coefficients as parameterized in Thompson et al.
(1987).
Asymmetry correction of (Finger et al., 1994)
Microstrain broadening by (Stephens, 1999).
#1(GU) = 0.000 #2(GV) = 0.000 #3(GW) = 10.448
#4(GP) = 0.000 #5(LX) = 2.798 #6(ptec) = 0.00
#7(trns) = 0.00 #8(shft) = 0.0000 #9(sfec) = 0.00
#10(S/L) = 0.0225 #11(H/L) = 0.0225 #12(eta) = 0.6000
#13(S400 ) = 7.2E-03 #14(S040 ) = 1.2E-02 #15(S004 ) = 1.9E-01
#16(S220 ) = -1.1E-03 #17(S202 ) = 3.5E-02 #18(S022 ) = 4.8E-02
#19(S301 ) = -1.7E-02 #20(S103 ) = -1.3E-02 #21(S121 ) = 1.0E-02
Peak tails are ignored where the intensity is below 0.0010 times the peak
Aniso. broadening axis 0.0 0.0 1.0
Stephens, P. W. (1999). J. Appl. Cryst. 32, 281–289.
Thompson, P., Cox, D. E. & Hastings, J. B. (1987). J. Appl.
Cryst.
20, 79–83. |
| Rp = 0.020 | 160 parameters |
| Rwp = 0.027 | 104 restraints |
| Rexp = 0.024 | H-atom parameters constrained |
| R(F2) = 0.03684 | (Δ/σ)max = 0.02 |
| χ2 = 1.346 | Background function: GSAS background function number 1 with 20 terms.
Shifted Chebyshev function of 1st kind
1: 1694.08 2: -1888.90 3: 780.704 4: -143.263
5: -45.4949 6: 51.0746 7: 7.48171 8: -51.4972
9: 12.7024 10: 17.5629 11: -17.3374 12: 2.64363
13: 3.91414 14: -2.81271 15: 4.80400 16: 5.62048
17: -5.95626 18: 1.07944 19: 1.90253 20: -5.42175 |
| ? data points | |
Crystal data top
| C13H28N4O2 | V = 1621.86 (7) Å3 |
| Mr = 272.39 | Z = 4 |
| Monoclinic, P21/n | Cu Kα1 radiation, λ = 1.5406 Å |
| a = 16.4453 (4) Å | µ = 0.61 mm−1 |
| b = 15.9587 (3) Å | T = 298 K |
| c = 6.20073 (11) Å | flat sheet, 7 × 7 mm |
| β = 94.7097 (13)° | |
Data collection top
Stoe STADI P
diffractometer | Scan method: step |
| Specimen mounting: drifted powder between two Mylar foils | 2θmin = 5.970°, 2θmax = 69.960°, 2θstep = 0.01° |
| Data collection mode: transmission | |
Refinement top
| Rp = 0.020 | ? data points |
| Rwp = 0.027 | 160 parameters |
| Rexp = 0.024 | 104 restraints |
| R(F2) = 0.03684 | H-atom parameters constrained |
| χ2 = 1.346 | |
Special details top
Experimental. The sample was ground lightly in a mortar, loaded between two Mylar foils and
fixed in the sample holder with a mask of 7.0 mm intrnal diameter. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| C1 | 0.2621 (3) | 0.2472 (3) | 0.0673 (7) | 0.034 (2)* | |
| C2 | 0.3255 (2) | 0.2898 (2) | 0.2283 (5) | 0.034 (2)* | |
| C3 | 0.2973 (2) | 0.1715 (2) | −0.0442 (6) | 0.034 (2)* | |
| C4 | 0.2023 (2) | 0.2112 (2) | 0.2237 (6) | 0.034 (2)* | |
| C5 | 0.2295 (2) | 0.3101 (2) | −0.1064 (6) | 0.034 (2)* | |
| N1 | 0.1251 (3) | 0.1791 (3) | 0.1229 (8) | 0.026 (2)* | |
| N2 | 0.1826 (3) | 0.3768 (3) | −0.0125 (8) | 0.026 (2)* | |
| C6 | 0.0647 (3) | 0.1662 (3) | 0.2803 (7) | 0.034 (2)* | |
| C7 | 0.1689 (3) | 0.4465 (3) | −0.1642 (7) | 0.034 (2)* | |
| C8 | −0.0163 (2) | 0.1410 (2) | 0.1551 (5) | 0.034 (2)* | |
| C9 | 0.1334 (2) | 0.5255 (2) | −0.0695 (6) | 0.034 (2)* | |
| C10 | 0.0887 (5) | 0.0963 (3) | 0.4438 (11) | 0.034 (2)* | |
| C11 | 0.1093 (4) | 0.4093 (6) | −0.3413 (11) | 0.034 (2)* | |
| C12 | 0.1049 (2) | 0.0075 (2) | 0.3684 (6) | 0.034 (2)* | |
| C13 | 0.0217 (2) | 0.3839 (2) | −0.3036 (6) | 0.034 (2)* | |
| N3 | 0.0934 (6) | 0.1332 (5) | 0.6256 (10) | 0.026 (2)* | |
| N4 | 0.1271 (5) | 0.4075 (6) | −0.5395 (11) | 0.026 (2)* | |
| O1 | 0.1129 (5) | 0.0690 (3) | 0.7776 (8) | 0.041 (3)* | |
| O2 | 0.0659 (3) | 0.3877 (5) | −0.7017 (9) | 0.041 (3)* | |
| H2A | 0.30562 | 0.34456 | 0.26736 | 0.05* | |
| H2B | 0.33426 | 0.25565 | 0.35766 | 0.05* | |
| H2C | 0.37643 | 0.29607 | 0.16202 | 0.05* | |
| H3A | 0.2533 | 0.14006 | −0.12066 | 0.05* | |
| H3B | 0.32584 | 0.13569 | 0.06336 | 0.05* | |
| H3C | 0.33488 | 0.19049 | −0.14639 | 0.05* | |
| H4A | 0.19005 | 0.25553 | 0.32522 | 0.05* | |
| H4B | 0.23006 | 0.16554 | 0.30608 | 0.05* | |
| H5A | 0.27567 | 0.33483 | −0.17413 | 0.05* | |
| H5B | 0.19427 | 0.28058 | −0.21664 | 0.05* | |
| H6A | 0.05703 | 0.21916 | 0.35931 | 0.05* | |
| H7A | 0.22082 | 0.46073 | −0.22594 | 0.05* | |
| H8A | −0.00644 | 0.12663 | 0.00722 | 0.05* | |
| H8B | −0.05444 | 0.18748 | 0.15434 | 0.05* | |
| H8C | −0.03912 | 0.09292 | 0.22474 | 0.05* | |
| H9A | 0.08214 | 0.51207 | −0.0104 | 0.05* | |
| H9B | 0.12415 | 0.56722 | −0.18263 | 0.05* | |
| H9C | 0.17152 | 0.54739 | 0.04432 | 0.05* | |
| H12A | 0.16304 | −0.00028 | 0.36057 | 0.05* | |
| H12B | 0.07677 | −0.00156 | 0.22685 | 0.05* | |
| H12C | 0.08509 | −0.03215 | 0.4703 | 0.05* | |
| H13A | −0.01637 | 0.42011 | −0.38727 | 0.05* | |
| H13B | 0.01354 | 0.38947 | −0.15118 | 0.05* | |
| H13C | 0.01267 | 0.32614 | −0.34844 | 0.05* | |
| H1N | 0.10579 | 0.21374 | 0.02342 | 0.05* | |
| H2N | 0.2088 | 0.39476 | 0.10631 | 0.05* | |
| H1O | 0.12593 | 0.08985 | 0.89653 | 0.05* | |
| H2O | 0.08686 | 0.37166 | −0.80967 | 0.05* | |
Geometric parameters (Å, º) top
| C1—C2 | 1.541 (5) | C7—C11 | 1.530 (5) |
| C1—C3 | 1.529 (5) | C7—H7A | 0.991 |
| C1—C4 | 1.547 (5) | C8—C6 | 1.538 (4) |
| C1—C5 | 1.536 (5) | C8—H8A | 0.9717 |
| C2—H2A | 0.9705 | C8—H8B | 0.9714 |
| C2—H2B | 0.9704 | C8—H8C | 0.9713 |
| C2—H2C | 0.9683 | C9—H9A | 0.9707 |
| C3—H3A | 0.9708 | C9—H9B | 0.9704 |
| C3—H3B | 0.9694 | C9—H9C | 0.9698 |
| C3—H3C | 0.9697 | C10—C12 | 1.522 (5) |
| C4—N1 | 1.462 (4) | C10—N3 | 1.269 (5) |
| C4—H4A | 0.979 | C11—C13 | 1.531 (5) |
| C4—H4B | 0.981 | C11—N4 | 1.287 (5) |
| C5—N2 | 1.464 (4) | C12—H12A | 0.9696 |
| C5—H5A | 0.980 | C12—H12B | 0.9688 |
| C5—H5B | 0.980 | C12—H12C | 0.9691 |
| N1—C6 | 1.463 (5) | C13—H13A | 0.9704 |
| N1—H1N | 0.869 | C13—H13B | 0.9697 |
| N2—C7 | 1.462 (5) | C13—H13C | 0.9714 |
| N2—H2N | 0.871 | N3—O1 | 1.411 (5) |
| C6—C8 | 1.538 (4) | N4—O2 | 1.400 (5) |
| C6—C10 | 1.537 (5) | O1—H1O | 0.821 |
| C6—H6A | 0.990 | O2—H2O | 0.819 |
| C7—C9 | 1.527 (4) | | |
| | | |
| C2—C1—C3 | 112.4 (3) | C10—C6—H6A | 109.30 |
| C2—C1—C4 | 100.9 (3) | N2—C7—C9 | 115.1 (4) |
| C2—C1—C5 | 110.2 (3) | N2—C7—C11 | 102.9 (5) |
| C3—C1—C4 | 105.9 (3) | N2—C7—H7A | 109.30 |
| C3—C1—C5 | 108.8 (3) | C9—C7—C11 | 110.7 (5) |
| C4—C1—C5 | 118.6 (4) | C9—C7—H7A | 109.10 |
| C1—C2—H2A | 109.60 | C11—C7—H7A | 109.50 |
| C1—C2—H2B | 109.55 | C6—C8—H8A | 109.53 |
| C1—C2—H2C | 109.45 | C6—C8—H8B | 109.59 |
| H2A—C2—H2B | 109.26 | C6—C8—H8C | 109.41 |
| H2A—C2—H2C | 109.48 | H8A—C8—H8B | 109.45 |
| H2B—C2—H2C | 109.49 | H8A—C8—H8C | 109.47 |
| C1—C3—H3A | 109.53 | H8B—C8—H8C | 109.37 |
| C1—C3—H3B | 109.51 | C7—C9—H9A | 109.54 |
| C1—C3—H3C | 109.43 | C7—C9—H9B | 109.36 |
| H3A—C3—H3B | 109.37 | C7—C9—H9C | 109.50 |
| H3A—C3—H3C | 109.39 | H9A—C9—H9B | 109.53 |
| H3B—C3—H3C | 109.59 | H9A—C9—H9C | 109.51 |
| C1—C4—N1 | 115.9 (4) | H9B—C9—H9C | 109.39 |
| C1—C4—H4A | 108.09 | C6—C10—C12 | 121.0 (4) |
| C1—C4—H4B | 108.07 | C6—C10—N3 | 104.0 (5) |
| N1—C4—H4A | 108.10 | C12—C10—N3 | 134.9 (6) |
| N1—C4—H4B | 108.20 | C7—C11—C13 | 123.2 (4) |
| H4A—C4—H4B | 108.30 | C7—C11—N4 | 120.7 (6) |
| C1—C5—N2 | 111.2 (4) | C13—C11—N4 | 115.5 (6) |
| C1—C5—H5A | 109.00 | C10—C12—H12A | 109.40 |
| C1—C5—H5B | 109.12 | C10—C12—H12B | 109.50 |
| N2—C5—H5A | 109.30 | C10—C12—H12C | 109.40 |
| N2—C5—H5B | 109.02 | H12A—C12—H12B | 109.56 |
| H5A—C5—H5B | 109.30 | H12A—C12—H12C | 109.55 |
| C4—N1—C6 | 112.2 (4) | H12B—C12—H12C | 109.41 |
| C4—N1—H1N | 109.50 | C11—C13—H13A | 109.60 |
| C6—N1—H1N | 109.60 | C11—C13—H13B | 109.41 |
| C5—N2—C7 | 110.8 (4) | C11—C13—H13C | 109.30 |
| C5—N2—H2N | 109.60 | H13A—C13—H13B | 109.36 |
| C7—N2—H2N | 109.50 | H13A—C13—H13C | 109.56 |
| N1—C6—C8 | 107.9 (3) | H13B—C13—H13C | 109.57 |
| N1—C6—C10 | 112.9 (5) | C10—N3—O1 | 104.5 (7) |
| N1—C6—H6A | 109.30 | C11—N4—O2 | 119.0 (7) |
| C8—C6—C10 | 108.0 (4) | N3—O1—H1O | 109.50 |
| C8—C6—H6A | 109.50 | N4—O2—H2O | 109.30 |
| | | |
| C6—N1—C4—C1 | −165.5 (4) | C2—C1—C5—N2 | −66.3 (4) |
| C4—N1—C6—C8 | 174.5 (3) | C5—C1—C4—N1 | 48.7 (5) |
| C4—N1—C6—C10 | −66.3 (5) | C3—C1—C4—N1 | −73.8 (4) |
| C5—N2—C7—C11 | 69.5 (5) | C2—C1—C4—N1 | 169.0 (3) |
| C7—N2—C5—C1 | 166.1 (4) | N1—C6—C10—C12 | −57.8 (7) |
| C5—N2—C7—C9 | −170.0 (4) | C8—C6—C10—C12 | 61.4 (7) |
| O1—N3—C10—C12 | −5.3 (14) | C8—C6—C10—N3 | −121.5 (7) |
| O1—N3—C10—C6 | 178.2 (6) | N1—C6—C10—N3 | 119.3 (7) |
| O2—N4—C11—C13 | 3.4 (13) | N2—C7—C11—C13 | 66.6 (8) |
| O2—N4—C11—C7 | −168.2 (7) | C9—C7—C11—N4 | 114.2 (8) |
| C3—C1—C5—N2 | 170.2 (3) | C9—C7—C11—C13 | −56.9 (8) |
| C4—C1—C5—N2 | 49.2 (5) | N2—C7—C11—N4 | −122.4 (9) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1O···N1i | 0.82 | 2.00 | 2.764 (7) | 154 |
| O2—H2O···N2ii | 0.82 | 2.10 | 2.835 (7) | 150 |
| Symmetry codes: (i) x, y, z+1; (ii) x, y, z−1. |
Experimental details
| | (Ialpha) | (Ibeta) |
| Crystal data |
| Chemical formula | C13H28N4O2 | C13H28N4O2 |
| Mr | 272.39 | 272.39 |
| Crystal system, space group | Triclinic, P1 | Monoclinic, P21/n |
| Temperature (K) | 298 | 298 |
| a, b, c (Å) | 15.3159 (3), 8.74371 (12), 6.21770 (9) | 16.4453 (4), 15.9587 (3), 6.20073 (11) |
| α, β, γ (°) | 81.1652 (10), 93.4835 (10), 96.4282 (8) | 90, 94.7097 (13), 90 |
| V (Å3) | 816.80 (2) | 1621.86 (7) |
| Z | 2 | 4 |
| Radiation type | Cu Kα1, λ = 1.5406 Å | Cu Kα1, λ = 1.5406 Å |
| µ (mm−1) | 0.61 | 0.61 |
| Specimen shape, size (mm) | Flat sheet, 7 × 7 | Flat sheet, 7 × 7 |
| |
| Data collection |
| Diffractometer | Stoe STADI P
diffractometer | Stoe STADI P
diffractometer |
| Specimen mounting | Drifted powder between two Mylar foils | Drifted powder between two Mylar foils |
| Data collection mode | Transmission | Transmission |
| Scan method | Step | Step |
| Absorption correction | ?
GSAS absorption/surface roughness correction (Larson & Von Dreele,
2004):
function number 4. Flat plate in transmission mode, absorption correction
Term (= MU.r/wave) = 0.16000
Correction is not refined. | – |
| Tmin, Tmax | 0.660, 0.669 | – |
| 2θ values (°) | 2θmin = 2.992 2θmax = 79.982 2θstep = 0.01 | 2θmin = 5.970 2θmax = 69.960 2θstep = 0.01 |
| |
| Refinement |
| R factors and goodness of fit | Rp = 0.017, Rwp = 0.023, Rexp = 0.019, R(F2) = 0.01476, χ2 = 1.416 | Rp = 0.020, Rwp = 0.027, Rexp = 0.024, R(F2) = 0.03684, χ2 = 1.346 |
| No. of data points | 7700 | ? |
| No. of parameters | 224 | 160 |
| No. of restraints | 104 | 104 |
| H-atom treatment | H-atom parameters constrained | H-atom parameters constrained |
Selected geometric parameters (Å, º) for (Ialpha) top| C4—N1 | 1.487 (5) | C7—C11 | 1.498 (5) |
| N1—C6 | 1.471 (5) | C10—N3 | 1.282 (5) |
| N2—C5 | 1.492 (4) | C11—N4 | 1.278 (5) |
| N2—C7 | 1.477 (5) | N3—O1 | 1.407 (5) |
| C6—C10 | 1.510 (5) | N4—O2 | 1.394 (5) |
| | | |
| N1—C6—C10 | 104.8 (5) | C7—C11—C13 | 118.7 (4) |
| N2—C7—C11 | 115.5 (5) | C7—C11—N4 | 116.2 (5) |
| C6—C10—C12 | 118.1 (4) | C13—C11—N4 | 123.9 (6) |
| C6—C10—N3 | 115.5 (5) | C10—N3—O1 | 112.3 (7) |
| C12—C10—N3 | 125.2 (6) | C11—N4—O2 | 109.7 (6) |
| | | |
| C6—N1—C4—C1 | 164.5 (3) | O2—N4—C11—C13 | −14.5 (10) |
| C4—N1—C6—C8 | −167.9 (3) | C8—C6—C10—C12 | −61.1 (7) |
| C7—N2—C5—C1 | −170.9 (3) | N1—C6—C10—N3 | −132.6 (7) |
| C5—N2—C7—C9 | 170.2 (3) | C9—C7—C11—C13 | 71.3 (6) |
| O1—N3—C10—C12 | −1.2 (11) | N2—C7—C11—N4 | 120.7 (6) |
Hydrogen-bond geometry (Å, º) for (Ialpha) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1O···N1i | 0.82 | 2.19 | 2.811 (8) | 133 |
| O2—H2O···N2ii | 0.82 | 2.17 | 2.834 (7) | 138 |
| Symmetry codes: (i) x, y, z−1; (ii) x, y, z+1. |
Selected geometric parameters (Å, º) for (Ibeta) top| C4—N1 | 1.462 (4) | C7—C11 | 1.530 (5) |
| C5—N2 | 1.464 (4) | C10—N3 | 1.269 (5) |
| N1—C6 | 1.463 (5) | C11—N4 | 1.287 (5) |
| N2—C7 | 1.462 (5) | N3—O1 | 1.411 (5) |
| C6—C10 | 1.537 (5) | N4—O2 | 1.400 (5) |
| | | |
| N1—C6—C10 | 112.9 (5) | C7—C11—C13 | 123.2 (4) |
| N2—C7—C11 | 102.9 (5) | C7—C11—N4 | 120.7 (6) |
| C6—C10—C12 | 121.0 (4) | C13—C11—N4 | 115.5 (6) |
| C6—C10—N3 | 104.0 (5) | C10—N3—O1 | 104.5 (7) |
| C12—C10—N3 | 134.9 (6) | C11—N4—O2 | 119.0 (7) |
| | | |
| C6—N1—C4—C1 | −165.5 (4) | O2—N4—C11—C13 | 3.4 (13) |
| C4—N1—C6—C8 | 174.5 (3) | C8—C6—C10—C12 | 61.4 (7) |
| C7—N2—C5—C1 | 166.1 (4) | N1—C6—C10—N3 | 119.3 (7) |
| C5—N2—C7—C9 | −170.0 (4) | C9—C7—C11—C13 | −56.9 (8) |
| O1—N3—C10—C12 | −5.3 (14) | N2—C7—C11—N4 | −122.4 (9) |
Hydrogen-bond geometry (Å, º) for (Ibeta) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1O···N1i | 0.82 | 2.00 | 2.764 (7) | 154 |
| O2—H2O···N2ii | 0.82 | 2.10 | 2.835 (7) | 150 |
| Symmetry codes: (i) x, y, z+1; (ii) x, y, z−1. |


 journal menu
journal menu organic compounds
organic compounds










![[Figure 1]](http://journals.iucr.org/c/issues/2010/09/00/sq3256/sq3256fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2010/09/00/sq3256/sq3256fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2010/09/00/sq3256/sq3256fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/c/issues/2010/09/00/sq3256/sq3256fig4thm.gif)




It is well known that both diastereomeric forms of hexamethyl propylene amine oxime, meso-HMPAO, (I), and d,l-HMPAO, (II), have important radiopharmaceutical applications in nuclear medicine after labelling with 99mTc (Neirinckx et al., 1987; Sasaki & Senda, 1997; Roth et al., 1992; Jurisson et al., 1986). For example, 99mTc-d,l-HMPAO has been widely used as a leukocyte labelling agent and for SPECT imaging of regional cerebral blood perfusion (Jurisson et al., 1986).
The most general reported synthetic route to the diastereomeric mixture of HMPAO involves two synthetic procedures (Jurisson et al., 1986; Banerjee et al., 1999), which give a 50:50 diastereomeric mixture of HMPAO, meso-HMPAO, (I), and d,l-HMPAO, (II), that can be separated by fractional crystallization using different solvents (Banerjee et al., 1999). 1H and 13C{1H} NMR spectroscopic techniques may be utilized to assess the relative amounts of meso-HMPAO, (I), and d,l-HMPAO, (II), isomers in the diastereomeric mixture and also to identify the purity of each isomer obtained from such a mixture (Feinstein-Jaffe et al., 1989; Babushkina et al., 2002). Although HMPAO has been used as a polydentate ligand to form metal–HMPAO complexes, some of which have been structurally characterized (Suksai et al., 2008), to the best of our knowledge there are no published reports of the molecular structure of free meso-HMPAO, (I), or d,l-HMPAO, (II). Since we are currently interested in preparing HMPAO for pharmaceutical applications, we generated the HMPAO mixture, isolated meso-HMPAO, and determined the structures of the two crystalline forms that resulted, (Iα) and (Iβ), by powder X-ray diffraction.
Both forms crystallize with one molecule in the asymmetric unit, having almost the same conformations for their corresponding atoms (Fig. 1) and RS/EE geometric isomerism (Table 1). The crystal system of form (Iα) is triclinic, space group P1, with two molecules in the unit cell, while (Iβ) is monoclinic, space group P21/n, with four molecules in the unit cell. In both structures, the molecules are in almost maximally extended forms, with the oxime groups apart. The C—C and N—C bond lengths and bond angles in the {C(HCH3)CNHCH2C(CH3)2CH2NHC(HCH3)C} unit are in their normal ranges (Allen et al., 1987) for single bonds and tetrahedral geometries (Tables 1 and 3). For each individual oxime group in the molecules of (Iα) and (Iβ), the mean distance between the O atom of the C═NOH group and the C atom of the attached CH3 group (2.67 Å) lies well within the sum of the van der Waals radii of O and C (3.2 Å; Bondi, 1964), indicating substantial intramolecular C···O interactions. The molecules of (Iα) and (Iβ) display similar bond distances and angles and intermolecular interactions to those in the closely related compound 3,3'-(trimethylenediamino)bis(3-methyl-2-butanone oxime) (C13H28N4O2; Hussain et al., 1984). The geometric data for (Iα) and (Iβ) are also comparable with those reported for the recently structurally characterized complex [Ni(meso-HMPAO)H].ClO4 (Suksai et al., 2008).
In the two phases, a major point of interest is the location of the CH3, C, N and O centres of each CH3C(NOH) unit in almost the same plane, as well as the short C—N bond lengths (average 1.30 Å), which readily indicate a Csp2═Nsp2 double bond. The N—O (ca 1.40 Å) bond lengths are also characteristic for an oxime group (Allen et al., 1987).
The molecules in both phases are joined by intermolecular O—H···N hydrogen bonds between the two oxime O—H groups of one molecule and the amine N atoms of two different adjacent molecules (Fig. 2, Tables 2 and 4), leading to one-dimensional chains along the [001] direction. The interactions between the chains for both phases involve short contacts between two methyl groups. In (Iα), this interaction is between the C8 methyl group on one chain and a corresponding C8ii [symmetry code: (ii) -x, -y, -z] methyl group in an adjacent chain (Fig. 3a), while for (Iβ) this interaction is between methyl groups C9 and C3iii [symmetry code: (iii) -x + 1/2, y + 1/2, -z +1/2] in adjacent chains. These C···C distances are 3.579 (4) Å for (Iα) and 3.593 (5) Å for (Iβ). In (Iα), the molecules of an individual chain are arranged in opposite orientations to the corresponding ones in an adjacent chain (as required by the inversion symmetry of the P1 space group), while in (Iβ) the molecules of one chain are rotated by 180° with respect to one another (as required by the two-fold screw axis symmetry along the b axis of the P21/n space group).
As expected, the measured melting points of the two phases are slightly different [424.6 K for (Iα) and 423.1 K for (Iβ)]. Although the difference is small, it may be due to slightly stronger interchain interactions for (Iα) than for (Iβ), which correlates with the marginally shorter interchain contact distances for (Iα).