The title compound, Na
+·3C
2H
6NO
2+·2SiF
62-·3C
2H
5NO
2, arose from an unexpected reaction of glycine and HF with the glass container. It is an unusual hybrid organic-inorganic network built up from chains of vertex-sharing NaF
4O
2 and SiF
6 octahedra. A pair of glycinium/glycine molecules bridges the chains into a sheet
via a centrosymmetric O

H
O link. The other organic species interact with the network by an extensive N-H
F hydrogen-bond network, including bifurcated and trifurcated bonds. Finally, an extremely short C-H
O interaction (H
O = 2.25 Å) is seen in the crystal structure. The Na atom has site symmetry
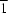
.
Supporting information
CCDC reference: 655491
An aqueous solution of glycine was mixed with excess 2 N hydrofluoric
acid. This solution was allowed to undergo evaporation by heating at 313 K.
The resulting solid was dried and purified by repeated crystallization in
water. The experiment was conducted in glass vessels. An aqueous saturated
solution of the compound synthesized as described above was prepared in water
at 323 K. The solution was filtered and kept to undergo slow evaporation by
cooling at a rate of 0.5 K per day. A portion of the mother solution was
poured into a Petri dish and allowed to undergo evaporation at room
temperature. The nucleation time was very long, but optically good tiny
crystals of (I) were seen in the Petri dish after about one month. These
crystals were used as seeds and suspended in the supersaturated mother
solution when the temperature reached 313 K. The growth temperature was
maintained at 313 K and large crystals of (I) of dimensions 10 × 6
× 3 mm were harvested after a week.
O-bound H atoms were located in difference maps and their positions were freely
refined, with Uiso(H) = 1.2Ueq(O). The other H atoms were
positioned geometrically, with C—H = 0.99 Å and N—H = 0.91 Å, and
refined as riding atoms, with Uiso(H) = 1.2Ueq(C,N).
The –NH3 groups were allowed to rotate but not to tip, to best fit the
electron density.
Data collection: COLLECT (Nonius, 1998); cell refinement: SCALEPACK (Otwinowski & Minor, 1997); data reduction: SCALEPACK, DENZO (Otwinowski & Minor, 1997) and SORTAV (Blessing,
1995); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEP-3 (Farrugia, 1997); software used to prepare material for publication: SHELXL97.
Sodium tris(glycinium) bis(hexafluorosilicate) tris(glycine)
top
Crystal data top
| Na+·3C2H6NO2+·2SiF62−·3C2H5NO2 | Z = 1 |
| Mr = 760.61 | F(000) = 390 |
| Triclinic, P1 | Dx = 1.901 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation, λ = 0.71073 Å |
| a = 5.5364 (1) Å | Cell parameters from 2981 reflections |
| b = 10.9918 (2) Å | θ = 1.0–27.5° |
| c = 11.8432 (2) Å | µ = 0.30 mm−1 |
| α = 107.631 (1)° | T = 120 K |
| β = 102.397 (1)° | Block, colourless |
| γ = 93.562 (1)° | 0.50 × 0.50 × 0.35 mm |
| V = 664.56 (2) Å3 | |
Data collection top
Nonius KappaCCD area-detector
diffractometer | 3041 independent reflections |
| Radiation source: fine-focus sealed tube | 2825 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.024 |
| ω and ϕ scans | θmax = 27.5°, θmin = 1.9° |
Absorption correction: multi-scan
(SADABS; Bruker, 2003) | h = −7→7 |
| Tmin = 0.863, Tmax = 0.901 | k = −14→14 |
| 14359 measured reflections | l = −15→15 |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: difmap and geom |
| R[F2 > 2σ(F2)] = 0.024 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.061 | w = 1/[σ2(Fo2) + (0.0238P)2 + 0.3675P]
where P = (Fo2 + 2Fc2)/3 |
| S = 1.07 | (Δ/σ)max = 0.002 |
| 3041 reflections | Δρmax = 0.39 e Å−3 |
| 212 parameters | Δρmin = −0.37 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97 (Sheldrick, 1997), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.043 (3) |
Crystal data top
| Na+·3C2H6NO2+·2SiF62−·3C2H5NO2 | γ = 93.562 (1)° |
| Mr = 760.61 | V = 664.56 (2) Å3 |
| Triclinic, P1 | Z = 1 |
| a = 5.5364 (1) Å | Mo Kα radiation |
| b = 10.9918 (2) Å | µ = 0.30 mm−1 |
| c = 11.8432 (2) Å | T = 120 K |
| α = 107.631 (1)° | 0.50 × 0.50 × 0.35 mm |
| β = 102.397 (1)° | |
Data collection top
Nonius KappaCCD area-detector
diffractometer | 3041 independent reflections |
Absorption correction: multi-scan
(SADABS; Bruker, 2003) | 2825 reflections with I > 2σ(I) |
| Tmin = 0.863, Tmax = 0.901 | Rint = 0.024 |
| 14359 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.024 | 0 restraints |
| wR(F2) = 0.061 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.07 | Δρmax = 0.39 e Å−3 |
| 3041 reflections | Δρmin = −0.37 e Å−3 |
| 212 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Na1 | 0.0000 | 0.0000 | 0.0000 | 0.00863 (14) | |
| Si1 | 0.38756 (5) | −0.03209 (3) | −0.24502 (3) | 0.00795 (9) | |
| F1 | 0.11609 (12) | −0.03242 (7) | −0.34021 (6) | 0.01558 (16) | |
| F2 | 0.52573 (12) | −0.06514 (6) | −0.36273 (6) | 0.01299 (15) | |
| F3 | 0.31163 (12) | −0.19241 (6) | −0.27208 (6) | 0.01305 (15) | |
| F4 | 0.65857 (13) | −0.03931 (7) | −0.14995 (6) | 0.01677 (16) | |
| F5 | 0.47129 (13) | 0.12586 (6) | −0.21366 (6) | 0.01395 (15) | |
| F6 | 0.25299 (14) | 0.00102 (7) | −0.12556 (6) | 0.01775 (16) | |
| C1 | 0.8232 (2) | 0.71694 (11) | 0.32627 (10) | 0.0132 (2) | |
| C2 | 1.0438 (2) | 0.76669 (11) | 0.28781 (10) | 0.0124 (2) | |
| H2A | 1.1231 | 0.6936 | 0.2460 | 0.015* | |
| H2B | 0.9865 | 0.8141 | 0.2302 | 0.015* | |
| N1 | 1.22650 (18) | 0.85362 (9) | 0.39816 (9) | 0.0135 (2) | |
| H1 | 1.3098 | 0.9147 | 0.3778 | 0.016* | |
| H2 | 1.3372 | 0.8070 | 0.4290 | 0.016* | |
| H3 | 1.1446 | 0.8925 | 0.4554 | 0.016* | |
| O1 | 0.78169 (16) | 0.77272 (8) | 0.42448 (7) | 0.01554 (18) | |
| O2 | 0.69211 (18) | 0.61321 (8) | 0.24422 (8) | 0.0194 (2) | |
| H4 | 0.569 (3) | 0.5640 (16) | 0.2747 (15) | 0.023* | |
| C3 | 0.2957 (2) | 0.50517 (11) | 0.38101 (10) | 0.0114 (2) | |
| C4 | 0.1459 (2) | 0.39720 (11) | 0.40237 (11) | 0.0128 (2) | |
| H4A | 0.1703 | 0.4145 | 0.4914 | 0.015* | |
| H4B | −0.0338 | 0.3958 | 0.3670 | 0.015* | |
| N2 | 0.21964 (18) | 0.26954 (9) | 0.34721 (9) | 0.01177 (19) | |
| H5 | 0.1301 | 0.2082 | 0.3645 | 0.014* | |
| H6 | 0.3853 | 0.2708 | 0.3784 | 0.014* | |
| H7 | 0.1888 | 0.2511 | 0.2648 | 0.014* | |
| O3 | 0.40409 (17) | 0.47046 (8) | 0.29384 (8) | 0.01905 (19) | |
| O4 | 0.29796 (16) | 0.61620 (8) | 0.44798 (8) | 0.01640 (18) | |
| C5 | −0.0559 (2) | 0.32435 (11) | 0.03744 (10) | 0.0103 (2) | |
| C6 | −0.3257 (2) | 0.34673 (11) | 0.02840 (10) | 0.0111 (2) | |
| H6A | −0.3332 | 0.4303 | 0.0892 | 0.013* | |
| H6B | −0.4014 | 0.3505 | −0.0540 | 0.013* | |
| N3 | −0.46791 (17) | 0.24179 (9) | 0.05096 (9) | 0.0120 (2) | |
| H8 | −0.6220 | 0.2626 | 0.0575 | 0.014* | |
| H9 | −0.3853 | 0.2307 | 0.1216 | 0.014* | |
| H10 | −0.4839 | 0.1675 | −0.0123 | 0.014* | |
| O5 | 0.01814 (15) | 0.23100 (8) | 0.06531 (8) | 0.01313 (17) | |
| O6 | 0.08726 (15) | 0.40592 (8) | 0.01570 (8) | 0.01578 (18) | |
| H11 | 0.0000 | 0.5000 | 0.0000 | 0.019* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Na1 | 0.0075 (3) | 0.0098 (3) | 0.0088 (3) | 0.0008 (2) | 0.0021 (2) | 0.0033 (2) |
| Si1 | 0.00734 (15) | 0.00913 (15) | 0.00725 (15) | 0.00048 (11) | 0.00165 (11) | 0.00271 (11) |
| F1 | 0.0102 (3) | 0.0158 (3) | 0.0183 (3) | 0.0027 (3) | −0.0019 (3) | 0.0055 (3) |
| F2 | 0.0160 (3) | 0.0137 (3) | 0.0112 (3) | 0.0029 (3) | 0.0072 (3) | 0.0040 (3) |
| F3 | 0.0141 (3) | 0.0098 (3) | 0.0150 (3) | −0.0002 (2) | 0.0025 (3) | 0.0048 (3) |
| F4 | 0.0110 (3) | 0.0227 (4) | 0.0155 (3) | −0.0006 (3) | −0.0032 (3) | 0.0095 (3) |
| F5 | 0.0171 (3) | 0.0097 (3) | 0.0140 (3) | −0.0008 (3) | 0.0049 (3) | 0.0022 (3) |
| F6 | 0.0207 (4) | 0.0185 (4) | 0.0156 (3) | −0.0001 (3) | 0.0121 (3) | 0.0029 (3) |
| C1 | 0.0184 (6) | 0.0107 (5) | 0.0121 (5) | 0.0022 (4) | 0.0040 (4) | 0.0058 (4) |
| C2 | 0.0156 (5) | 0.0120 (5) | 0.0096 (5) | 0.0010 (4) | 0.0033 (4) | 0.0033 (4) |
| N1 | 0.0148 (5) | 0.0132 (5) | 0.0123 (5) | 0.0011 (4) | 0.0027 (4) | 0.0045 (4) |
| O1 | 0.0203 (4) | 0.0149 (4) | 0.0111 (4) | −0.0009 (3) | 0.0058 (3) | 0.0031 (3) |
| O2 | 0.0278 (5) | 0.0136 (4) | 0.0139 (4) | −0.0070 (4) | 0.0085 (4) | −0.0001 (3) |
| C3 | 0.0097 (5) | 0.0124 (5) | 0.0119 (5) | 0.0009 (4) | −0.0001 (4) | 0.0057 (4) |
| C4 | 0.0141 (5) | 0.0093 (5) | 0.0164 (5) | 0.0023 (4) | 0.0069 (4) | 0.0039 (4) |
| N2 | 0.0121 (4) | 0.0105 (5) | 0.0139 (5) | 0.0023 (3) | 0.0048 (4) | 0.0044 (4) |
| O3 | 0.0266 (5) | 0.0149 (4) | 0.0156 (4) | −0.0044 (3) | 0.0103 (4) | 0.0028 (3) |
| O4 | 0.0179 (4) | 0.0103 (4) | 0.0216 (4) | 0.0019 (3) | 0.0066 (3) | 0.0047 (3) |
| C5 | 0.0112 (5) | 0.0101 (5) | 0.0085 (5) | 0.0008 (4) | 0.0020 (4) | 0.0019 (4) |
| C6 | 0.0106 (5) | 0.0105 (5) | 0.0132 (5) | 0.0020 (4) | 0.0034 (4) | 0.0050 (4) |
| N3 | 0.0091 (4) | 0.0137 (5) | 0.0137 (5) | 0.0006 (4) | 0.0028 (3) | 0.0054 (4) |
| O5 | 0.0114 (4) | 0.0102 (4) | 0.0193 (4) | 0.0030 (3) | 0.0037 (3) | 0.0068 (3) |
| O6 | 0.0126 (4) | 0.0159 (4) | 0.0230 (4) | 0.0011 (3) | 0.0055 (3) | 0.0118 (3) |
Geometric parameters (Å, º) top
| Na1—F4i | 2.2150 (6) | O2—H4 | 1.031 (17) |
| Na1—F4ii | 2.2150 (6) | C3—O4 | 1.2353 (14) |
| Na1—F6iii | 2.2529 (7) | C3—O3 | 1.2759 (14) |
| Na1—F6 | 2.2529 (7) | C3—C4 | 1.5197 (15) |
| Na1—O5iii | 2.4085 (8) | C4—N2 | 1.4809 (14) |
| Na1—O5 | 2.4085 (8) | C4—H4A | 0.9900 |
| Si1—F1 | 1.6730 (7) | C4—H4B | 0.9900 |
| Si1—F5 | 1.6741 (7) | N2—H5 | 0.9100 |
| Si1—F2 | 1.6871 (7) | N2—H6 | 0.9100 |
| Si1—F6 | 1.6922 (7) | N2—H7 | 0.9100 |
| Si1—F4 | 1.6938 (7) | O3—H4 | 1.435 (17) |
| Si1—F3 | 1.7008 (7) | C5—O5 | 1.2365 (14) |
| F4—Na1i | 2.2150 (6) | C5—O6 | 1.2797 (14) |
| C1—O1 | 1.2175 (14) | C5—C6 | 1.5152 (15) |
| C1—O2 | 1.3017 (14) | C6—N3 | 1.4786 (14) |
| C1—C2 | 1.5179 (16) | C6—H6A | 0.9900 |
| C2—N1 | 1.4833 (14) | C6—H6B | 0.9900 |
| C2—H2A | 0.9900 | N3—H8 | 0.9100 |
| C2—H2B | 0.9900 | N3—H9 | 0.9100 |
| N1—H1 | 0.9100 | N3—H10 | 0.9100 |
| N1—H2 | 0.9100 | O6—H11 | 1.22 |
| N1—H3 | 0.9100 | | |
| | | |
| F4i—Na1—F4ii | 180.0 | C2—N1—H1 | 109.5 |
| F4i—Na1—F6iii | 92.65 (3) | C2—N1—H2 | 109.5 |
| F4ii—Na1—F6iii | 87.35 (3) | H1—N1—H2 | 109.5 |
| F4i—Na1—F6 | 87.35 (3) | C2—N1—H3 | 109.5 |
| F4ii—Na1—F6 | 92.65 (3) | H1—N1—H3 | 109.5 |
| F6iii—Na1—F6 | 180.0 | H2—N1—H3 | 109.5 |
| F4i—Na1—O5iii | 95.82 (3) | C1—O2—H4 | 115.0 (9) |
| F4ii—Na1—O5iii | 84.18 (3) | O4—C3—O3 | 126.98 (11) |
| F6iii—Na1—O5iii | 92.76 (3) | O4—C3—C4 | 117.35 (10) |
| F6—Na1—O5iii | 87.24 (3) | O3—C3—C4 | 115.66 (10) |
| F4i—Na1—O5 | 84.18 (3) | N2—C4—C3 | 112.29 (9) |
| F4ii—Na1—O5 | 95.82 (3) | N2—C4—H4A | 109.1 |
| F6iii—Na1—O5 | 87.24 (3) | C3—C4—H4A | 109.1 |
| F6—Na1—O5 | 92.76 (3) | N2—C4—H4B | 109.1 |
| O5iii—Na1—O5 | 180.0 | C3—C4—H4B | 109.1 |
| F1—Si1—F5 | 92.75 (4) | H4A—C4—H4B | 107.9 |
| F1—Si1—F2 | 90.01 (4) | C4—N2—H5 | 109.5 |
| F5—Si1—F2 | 90.28 (3) | C4—N2—H6 | 109.5 |
| F1—Si1—F6 | 90.82 (4) | H5—N2—H6 | 109.5 |
| F5—Si1—F6 | 89.69 (4) | C4—N2—H7 | 109.5 |
| F2—Si1—F6 | 179.16 (4) | H5—N2—H7 | 109.5 |
| F1—Si1—F4 | 177.28 (4) | H6—N2—H7 | 109.5 |
| F5—Si1—F4 | 89.97 (4) | C3—O3—H4 | 119.5 (7) |
| F2—Si1—F4 | 90.02 (4) | O5—C5—O6 | 122.74 (10) |
| F6—Si1—F4 | 89.14 (4) | O5—C5—C6 | 120.26 (10) |
| F1—Si1—F3 | 89.44 (3) | O6—C5—C6 | 117.01 (10) |
| F5—Si1—F3 | 177.75 (4) | N3—C6—C5 | 110.35 (9) |
| F2—Si1—F3 | 90.26 (3) | N3—C6—H6A | 109.6 |
| F6—Si1—F3 | 89.74 (4) | C5—C6—H6A | 109.6 |
| F4—Si1—F3 | 87.84 (4) | N3—C6—H6B | 109.6 |
| Si1—F4—Na1i | 165.79 (5) | C5—C6—H6B | 109.6 |
| Si1—F6—Na1 | 164.78 (4) | H6A—C6—H6B | 108.1 |
| O1—C1—O2 | 126.18 (11) | C6—N3—H8 | 109.5 |
| O1—C1—C2 | 120.92 (10) | C6—N3—H9 | 109.5 |
| O2—C1—C2 | 112.90 (10) | H8—N3—H9 | 109.5 |
| N1—C2—C1 | 108.84 (9) | C6—N3—H10 | 109.5 |
| N1—C2—H2A | 109.9 | H8—N3—H10 | 109.5 |
| C1—C2—H2A | 109.9 | H9—N3—H10 | 109.5 |
| N1—C2—H2B | 109.9 | C5—O5—Na1 | 145.55 (7) |
| C1—C2—H2B | 109.9 | C5—O6—H11 | 117 |
| H2A—C2—H2B | 108.3 | | |
| | | |
| F5—Si1—F4—Na1i | 33.61 (17) | O1—C1—C2—N1 | 19.66 (15) |
| F2—Si1—F4—Na1i | 123.89 (17) | O2—C1—C2—N1 | −160.74 (10) |
| F6—Si1—F4—Na1i | −56.08 (17) | O4—C3—C4—N2 | −161.49 (10) |
| F3—Si1—F4—Na1i | −145.85 (17) | O3—C3—C4—N2 | 19.24 (14) |
| F1—Si1—F6—Na1 | 51.34 (17) | O5—C5—C6—N3 | −2.66 (14) |
| F5—Si1—F6—Na1 | 144.08 (17) | O6—C5—C6—N3 | 177.66 (9) |
| F4—Si1—F6—Na1 | −125.94 (17) | O6—C5—O5—Na1 | −110.52 (13) |
| F3—Si1—F6—Na1 | −38.10 (17) | C6—C5—O5—Na1 | 69.82 (17) |
| F4i—Na1—F6—Si1 | 138.38 (17) | F4i—Na1—O5—C5 | 163.02 (13) |
| F4ii—Na1—F6—Si1 | −41.62 (17) | F4ii—Na1—O5—C5 | −16.98 (13) |
| O5iii—Na1—F6—Si1 | 42.42 (17) | F6iii—Na1—O5—C5 | −104.03 (13) |
| O5—Na1—F6—Si1 | −137.58 (17) | F6—Na1—O5—C5 | 75.97 (13) |
| Symmetry codes: (i) −x+1, −y, −z; (ii) x−1, y, z; (iii) −x, −y, −z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···F2iv | 0.91 | 1.91 | 2.8115 (12) | 169 |
| N1—H1···F5iv | 0.91 | 2.45 | 3.0735 (12) | 126 |
| N1—H2···O4v | 0.91 | 2.18 | 2.8769 (13) | 133 |
| N1—H2···O1v | 0.91 | 2.52 | 3.2299 (13) | 135 |
| N1—H3···F1vi | 0.91 | 2.35 | 3.1782 (12) | 151 |
| O2—H4···O3 | 1.031 (17) | 1.435 (17) | 2.4495 (12) | 166.7 (16) |
| N2—H5···F1iii | 0.91 | 2.20 | 3.0750 (12) | 161 |
| N2—H5···O1vii | 0.91 | 2.38 | 2.8803 (13) | 115 |
| N2—H5···F3iii | 0.91 | 2.43 | 2.8760 (12) | 111 |
| N2—H6···F2i | 0.91 | 2.30 | 2.7571 (12) | 110 |
| N2—H6···O4vii | 0.91 | 2.34 | 3.0904 (13) | 140 |
| N2—H6···F3i | 0.91 | 2.36 | 3.0088 (12) | 128 |
| N2—H7···O5 | 0.91 | 2.28 | 3.1710 (13) | 165 |
| N2—H7···F4i | 0.91 | 2.61 | 3.1022 (12) | 115 |
| N3—H8···O5ii | 0.91 | 2.03 | 2.8840 (12) | 156 |
| N3—H8···O6ii | 0.91 | 2.40 | 3.1720 (13) | 143 |
| N3—H9···F3iii | 0.91 | 1.92 | 2.8014 (12) | 163 |
| N3—H10···F6ii | 0.91 | 2.15 | 2.9180 (12) | 142 |
| N3—H10···F5ii | 0.91 | 2.24 | 2.9383 (12) | 133 |
| O6—H11···O6viii | 1.22 | 1.22 | 2.4309 (16) | 180 |
| C6—H6A···O2ii | 0.99 | 2.25 | 3.2316 (14) | 172 |
| Symmetry codes: (i) −x+1, −y, −z; (ii) x−1, y, z; (iii) −x, −y, −z; (iv) −x+2, −y+1, −z; (v) x+1, y, z; (vi) x+1, y+1, z+1; (vii) −x+1, −y+1, −z+1; (viii) −x, −y+1, −z. |
Experimental details
| Crystal data |
| Chemical formula | Na+·3C2H6NO2+·2SiF62−·3C2H5NO2 |
| Mr | 760.61 |
| Crystal system, space group | Triclinic, P1 |
| Temperature (K) | 120 |
| a, b, c (Å) | 5.5364 (1), 10.9918 (2), 11.8432 (2) |
| α, β, γ (°) | 107.631 (1), 102.397 (1), 93.562 (1) |
| V (Å3) | 664.56 (2) |
| Z | 1 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.30 |
| Crystal size (mm) | 0.50 × 0.50 × 0.35 |
| |
| Data collection |
| Diffractometer | Nonius KappaCCD area-detector
diffractometer |
| Absorption correction | Multi-scan
(SADABS; Bruker, 2003) |
| Tmin, Tmax | 0.863, 0.901 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 14359, 3041, 2825 |
| Rint | 0.024 |
| (sin θ/λ)max (Å−1) | 0.650 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.024, 0.061, 1.07 |
| No. of reflections | 3041 |
| No. of parameters | 212 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.39, −0.37 |
Selected geometric parameters (Å, º) top| Na1—F4i | 2.2150 (6) | Si1—F2 | 1.6871 (7) |
| Na1—F6 | 2.2529 (7) | Si1—F6 | 1.6922 (7) |
| Na1—O5 | 2.4085 (8) | Si1—F4 | 1.6938 (7) |
| Si1—F1 | 1.6730 (7) | Si1—F3 | 1.7008 (7) |
| Si1—F5 | 1.6741 (7) | | |
| | | |
| Si1—F4—Na1ii | 165.79 (5) | Si1—F6—Na1 | 164.78 (4) |
| | | |
| O1—C1—C2—N1 | 19.66 (15) | O5—C5—C6—N3 | −2.66 (14) |
| O3—C3—C4—N2 | 19.24 (14) | | |
| Symmetry codes: (i) x−1, y, z; (ii) −x+1, −y, −z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···F2iii | 0.91 | 1.91 | 2.8115 (12) | 169 |
| N1—H1···F5iii | 0.91 | 2.45 | 3.0735 (12) | 126 |
| N1—H2···O4iv | 0.91 | 2.18 | 2.8769 (13) | 133 |
| N1—H2···O1iv | 0.91 | 2.52 | 3.2299 (13) | 135 |
| N1—H3···F1v | 0.91 | 2.35 | 3.1782 (12) | 151 |
| O2—H4···O3 | 1.031 (17) | 1.435 (17) | 2.4495 (12) | 166.7 (16) |
| N2—H5···F1vi | 0.91 | 2.20 | 3.0750 (12) | 161 |
| N2—H5···O1vii | 0.91 | 2.38 | 2.8803 (13) | 115 |
| N2—H5···F3vi | 0.91 | 2.43 | 2.8760 (12) | 111 |
| N2—H6···F2ii | 0.91 | 2.30 | 2.7571 (12) | 110 |
| N2—H6···O4vii | 0.91 | 2.34 | 3.0904 (13) | 140 |
| N2—H6···F3ii | 0.91 | 2.36 | 3.0088 (12) | 128 |
| N2—H7···O5 | 0.91 | 2.28 | 3.1710 (13) | 165 |
| N2—H7···F4ii | 0.91 | 2.61 | 3.1022 (12) | 115 |
| N3—H8···O5i | 0.91 | 2.03 | 2.8840 (12) | 156 |
| N3—H8···O6i | 0.91 | 2.40 | 3.1720 (13) | 143 |
| N3—H9···F3vi | 0.91 | 1.92 | 2.8014 (12) | 163 |
| N3—H10···F6i | 0.91 | 2.15 | 2.9180 (12) | 142 |
| N3—H10···F5i | 0.91 | 2.24 | 2.9383 (12) | 133 |
| O6—H11···O6viii | 1.22 | 1.22 | 2.4309 (16) | 180 |
| C6—H6A···O2i | 0.99 | 2.25 | 3.2316 (14) | 172 |
| Symmetry codes: (i) x−1, y, z; (ii) −x+1, −y, −z; (iii) −x+2, −y+1, −z; (iv) x+1, y, z; (v) x+1, y+1, z+1; (vi) −x, −y, −z; (vii) −x+1, −y+1, −z+1; (viii) −x, −y+1, −z. |


 journal menu
journal menu metal-organic compounds
metal-organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2007/07/00/sq3076/sq3076fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2007/07/00/sq3076/sq3076fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2007/07/00/sq3076/sq3076fig3thm.gif)




The title compound, (I), Na(SiF6)2(C2H6NO2)3(C2H5NO2)3, arose as an unexpected product during our studies of amino acid fluorides as possible non-linear optical materials. The Na and Si were presumably incorporated from the walls of the glass reaction vessel, mediated by reaction with hydrofluoric acid.
The structure of (I) (Fig. 1) is built up from trans-NaF4O2 octahedra (Na site symmetry 1) and SiF6 octahedra, sharing corners via atoms F4 and F6. The geometries of the Na and Si atoms (Table 1) may be regarded as normal (Allen et al., 1995). Considered by themselves, they form chains propagating in [100] with each Na octahedron linked to its neighbour by two Si polyhedra via F atoms, with notably large Na—F—Si bond angles greater than 164°. The square formed by atoms Na1, Si1, Na1i and Si1i [symmetry code: (i) 1 - x, -y, -z] is constrained to be exactly flat by symmetry and the Na···Si1···Na1i [90.58 (3)°] and Si1···Na1···Si1i [89.42 (3)°] angles barely deviate from 90°. The NaF4O2 polyhedron is a rare one, with only two other examples recorded in Version 5.27 of the Cambridge Structural Database (Allen, 2002), in structures otherwise unrelated to (I).
There are three organic molecules in the asymmetric unit of (I). The C1 species is a well defined C2H6NO2+ glycinium cation with C1—O2 [1.3018 (14) Å] much longer than C1—O1 [1.2175 (14) Å], indicating localized single and double bonds, respectively. The positive charge of the cation is localized on the N atom.
The C2 spcies is a neutral C2H5NO2 glycine molecule in its normal (Marsh, 1958) zwitterionic form (i.e. nominal H-atomtransfer from O3 or O4 to N2). Consequently, the C3—O3 and C3—O4 bond lengths [1.2759 (14) and 1.2354 (14) Å, respectively] approach each other, implying a degree of delocalization. The C1 and C3 molecules are linked by an O2—H4···O3 hydrogen bond (Table 2) and it is possible that C3—O3 is lengthened relative to C3—O4 because atom O3 accepts this short strong bond, with O···O = 2.4495 (12) Å.
The C5 molecule sits close to an inversion centre, resulting in an O6···O6ii contact of 2.4312 (16) Å [symmetry code: (ii) -x, 1 - y, -z], whereas atom O5 makes a bond to an adjacent Na+ cation [C5—O5 = 1.2366 (14) and C5—O6 = 1.2796 (14) Å]. In order to achieve overall change balance in the structure and to justify the very short O···O contact, we have assumed a symmetrical (single potential well) hydrogen bond (Ichikawa, 1978), with atom H11 located on the inversion centre mid-way between the two crystallorgaphically equivalent O6 atoms. This is strongly supported by our refinements of the structure in space group P1, which resulted in a significant difference peak almost mid-way between the two non-equivalent O atoms in the non-centrosymmetric model. Even so, in the centrosymmetric model, when Uiso(H11) was allowed to refine freely, a value of 0.062 (9) Å2 resulted, significantly higher than those of the other H atoms, perhaps suggesting disorder.
Thus, based on the present data, a situation of disordered asymmetric O—H···O and O···H—O bonds (double potential well) cannot be completely ruled out, as they are known to be hard to distinguish from X-ray data alone and indeed may be temperature dependent (Wilson, 2001). Either way, this ensemble in (I) has a formula of C4H11N2O4, i.e. equivalent to one glycinium cation and one glycine molecule (C2H6NO2+ + C2H5NO2). These glycinium/glycine pairs serve to bridge the [100] inorganic chains into (001) sheets (Fig. 2). For all three organic molecules, the N atom of the –NH3 group and one of the O atoms of the –CO2 group are close to being eclipsed (Table 1), which is not an uncommon conformation for glycine (Natarajan & Zangrando, 1992). The co-existence of glycine molecules and glycinium cations in the same structure is also very common (Nemec et al., 1998).
A large number of N—H···F hydrogen bonds (Table 2) help to consolidate the crystal packing of (I). It is notable that some of these are bifurcated N—H···(F,F) bonds and some are trifurcated N—H···(F,F,F) links. Finally, an exceptionally short C6–H6A···O2(x - 1, y z) interaction (Taylor & Kennard, 1982) with H···O = 2.25 Å occurs in the structure of (I), although its role in the complex crystal packing is not clear.
Together, these interactions generate a structure in which the (001) hybrid sheets sandwich the C1 and C2 organic molecules, with a dense mesh of N—H···F hydrogen bonds linking the two together (Fig. 3).