
It is demonstrated that H atoms can be located by the spectroscopic method of deuteron NMR. The requirement is that the `heavy-atom' positions are known from diffraction studies. The technique allows an accuracy of the order of 0.01 Å. The compound studied is ammonium persulphate (APS), (NH

)

S
O

. APS crystallizes in space group
P2

/

with lattice parameters
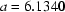
(2),
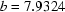
(3),
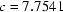
(3) Å and
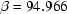
(1)° at
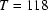
K. In perdeuterated crystals of APS, only one of the deuterons of every ND
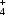
ion becomes localized at low temperatures. Therefore, most of this work uses samples with 9% deuteration. In such crystals, most of the ammonium ions containing deuterons come in the form of NDH
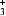
ions. At
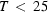
K, the single deuteron of these ions becomes localized in one of four equilibrium sites. The deuteron site occupancies differ from each other and are measured at 17 K. The deuterons are located in three steps. (i) The deuteron quadrupole-coupling (QC) tensors are measured at 17 K. Their unique principal directions are identified, as is well justified, with the N-D bond directions. (ii) The fine structure of a deuteron NMR line is analyzed in terms of the magnetic dipole-dipole interactions between all nuclei in an NDH
ion to obtain the N-D and D-H internuclear distances. (iii) An empirical relation between deuteron QC constants and D
O distances in N-D
O hydrogen bonds is exploited to assign the N-D bond vectors to the appropriate N atom of which there are four in the unit cell. The results are highly relevant for an understanding of the complex tunnelling and stochastic reorientation dynamics of the ammonium ions in APS. They are verified by a complementary X-ray diffraction study.
Supporting information
Data collection: Collect Package, BRUKER NONIUS B.V. 1999; cell refinement: Collect Package, BRUKER NONIUS B.V. 1999; data reduction: Collect Package, BRUKER NONIUS B.V. 1999; program(s) used to solve structure: SHELXS97, Sheldrick 1990; program(s) used to refine structure: SHELXL97, Sheldrick 1997; molecular graphics: SCHAKAL 99, Keller 1999.
ammonium persulfate-d8
top
Crystal data top
| (SO4)2(NH4)2 | F(000) = 236 |
| Mr = 228.20 | Dx = 2.016 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| a = 6.1340 (2) Å | Cell parameters from 786 reflections |
| b = 7.9324 (3) Å | θ = 1.0–27.9° |
| c = 7.7541 (3) Å | µ = 0.73 mm−1 |
| β = 94.966 (1)° | T = 118 K |
| V = 375.88 (2) Å3 | Fragment, colourless |
| Z = 2 | 0.32 × 0.20 × 0.20 mm |
Data collection top
NONIUS KappaCCD diffraktometer
diffractometer | 768 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.024 |
| Graphite monochromator | θmax = 27.9°, θmin = 5.1° |
| phi– and ω scans | h = −8→8 |
| 1393 measured reflections | k = −10→10 |
| 791 independent reflections | l = −10→10 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.028 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.069 | Refined isotropic |
| S = 1.07 | w = 1/[σ2(Fo2) + (0.0151P)2 + 0.2285P]
where P = (Fo2 + 2Fc2)/3 |
| 791 reflections | (Δ/σ)max = 0.002 |
| 71 parameters | Δρmax = 0.25 e Å−3 |
| 0 restraints | Δρmin = −0.30 e Å−3 |
Crystal data top
| (SO4)2(NH4)2 | V = 375.88 (2) Å3 |
| Mr = 228.20 | Z = 2 |
| Monoclinic, P21/n | Mo Kα radiation |
| a = 6.1340 (2) Å | µ = 0.73 mm−1 |
| b = 7.9324 (3) Å | T = 118 K |
| c = 7.7541 (3) Å | 0.32 × 0.20 × 0.20 mm |
| β = 94.966 (1)° | |
Data collection top
NONIUS KappaCCD diffraktometer
diffractometer | 768 reflections with I > 2σ(I) |
| 1393 measured reflections | Rint = 0.024 |
| 791 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.028 | 0 restraints |
| wR(F2) = 0.069 | Refined isotropic |
| S = 1.07 | Δρmax = 0.25 e Å−3 |
| 791 reflections | Δρmin = −0.30 e Å−3 |
| 71 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| S1 | 0.21386 (5) | 0.85461 (4) | 0.63745 (4) | 0.01348 (16) | |
| O3 | 0.11829 (13) | 1.01867 (13) | 0.52348 (13) | 0.0213 (3) | |
| O1 | 0.18431 (17) | 0.71093 (13) | 0.52550 (13) | 0.0242 (3) | |
| O4 | 0.08329 (16) | 0.84816 (12) | 0.78417 (12) | 0.0191 (2) | |
| O2 | 0.43508 (14) | 0.91084 (13) | 0.67523 (13) | 0.0198 (2) | |
| N1 | 0.2287 (2) | 0.88530 (17) | 0.14819 (16) | 0.0172 (3) | |
| D1 | 0.217 (4) | 0.862 (2) | 0.042 (3) | 0.036 (6)* | |
| D2 | 0.352 (4) | 0.908 (3) | 0.182 (3) | 0.036 (5)* | |
| D3 | 0.191 (4) | 0.798 (3) | 0.196 (3) | 0.048 (7)* | |
| D4 | 0.143 (3) | 0.963 (3) | 0.169 (2) | 0.022 (4)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| S1 | 0.0126 (2) | 0.0144 (2) | 0.0133 (2) | 0.00095 (10) | 0.00006 (13) | −0.00041 (10) |
| O3 | 0.0098 (5) | 0.0242 (5) | 0.0285 (6) | −0.0041 (4) | −0.0056 (4) | 0.0105 (4) |
| O1 | 0.0247 (5) | 0.0233 (6) | 0.0247 (5) | −0.0002 (4) | 0.0030 (4) | −0.0107 (4) |
| O4 | 0.0188 (5) | 0.0248 (5) | 0.0141 (5) | 0.0022 (4) | 0.0028 (4) | 0.0008 (3) |
| O2 | 0.0130 (4) | 0.0217 (5) | 0.0239 (5) | 0.0003 (4) | −0.0028 (4) | 0.0016 (4) |
| N1 | 0.0161 (6) | 0.0173 (6) | 0.0181 (6) | 0.0005 (4) | 0.0002 (5) | −0.0002 (4) |
Geometric parameters (Å, º) top
| S1—O2 | 1.4344 (9) | N1—D1 | 0.84 (2) |
| S1—O1 | 1.4346 (10) | N1—D2 | 0.80 (2) |
| S1—O4 | 1.4478 (10) | N1—D3 | 0.83 (3) |
| S1—O3 | 1.6518 (10) | N1—D4 | 0.84 (2) |
| O3—O3i | 1.4949 (17) | | |
| | | |
| O2—S1—O1 | 116.00 (6) | D1—N1—D2 | 112 (2) |
| O2—S1—O4 | 115.36 (6) | D1—N1—D3 | 104 (2) |
| O1—S1—O4 | 113.45 (6) | D2—N1—D3 | 110 (2) |
| O2—S1—O3 | 98.62 (5) | D1—N1—D4 | 110.0 (19) |
| O1—S1—O3 | 106.37 (6) | D2—N1—D4 | 111.2 (19) |
| O4—S1—O3 | 104.68 (5) | D3—N1—D4 | 109 (2) |
| O3i—O3—S1 | 105.44 (9) | | |
| | | |
| O2—S1—O3—O3i | −178.60 (10) | O4—S1—O3—O3i | −59.42 (11) |
| O1—S1—O3—O3i | 60.96 (11) | | |
| Symmetry code: (i) −x, −y+2, −z+1. |
Experimental details
| Crystal data |
| Chemical formula | (SO4)2(NH4)2 |
| Mr | 228.20 |
| Crystal system, space group | Monoclinic, P21/n |
| Temperature (K) | 118 |
| a, b, c (Å) | 6.1340 (2), 7.9324 (3), 7.7541 (3) |
| β (°) | 94.966 (1) |
| V (Å3) | 375.88 (2) |
| Z | 2 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.73 |
| Crystal size (mm) | 0.32 × 0.20 × 0.20 |
| |
| Data collection |
| Diffractometer | NONIUS KappaCCD diffraktometer
diffractometer |
| Absorption correction | – |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 1393, 791, 768 |
| Rint | 0.024 |
| (sin θ/λ)max (Å−1) | 0.658 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.028, 0.069, 1.07 |
| No. of reflections | 791 |
| No. of parameters | 71 |
| H-atom treatment | Refined isotropic |
| Δρmax, Δρmin (e Å−3) | 0.25, −0.30 |


 journal menu
journal menu research papers
research papers










![[Figure 1]](http://journals.iucr.org/b/issues/2002/05/00/sn0024/sn0024fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/b/issues/2002/05/00/sn0024/sn0024fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/b/issues/2002/05/00/sn0024/sn0024fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/b/issues/2002/05/00/sn0024/sn0024fig4thm.gif)
![[Figure 5]](http://journals.iucr.org/b/issues/2002/05/00/sn0024/sn0024fig5thm.gif)
![[Figure 6]](http://journals.iucr.org/b/issues/2002/05/00/sn0024/sn0024fig6thm.gif)
![[Figure 7]](http://journals.iucr.org/b/issues/2002/05/00/sn0024/sn0024fig7thm.gif)
![[Figure 8]](http://journals.iucr.org/b/issues/2002/05/00/sn0024/sn0024fig8thm.gif)



