In the title compound, C
8H
12N
+·C
2H
4O
5P
-, the anions are linked by two O-H

O hydrogen bonds [H
O both 1.75 Å, O
O = 2.5781 (15) and 2.5834 (15) Å, and O-H
O = 169 and 176°] into sheets built from alternating
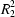
(8) and
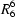
(32) rings. Each cation is linked to an anion sheet by three N-H
O hydrogen bonds [H
O = 1.88-2.04 Å, N
O = 2.7603 (16)-2.9334 (17) Å and N-H
O = 162-166°], such that all the cations pendent from one face of the sheet are of the
R configuration, while all those pendent from the opposite face are of the
S configuration.
Supporting information
CCDC reference: 235338
Equimolar quantities of racemic 2-phenylethylamine and carboxymethylphosphonic acid were dissolved separately in methanol The solutions were mixed and the mixture was then set aside to crystallize, providing analytically pure (I). Analysis found: C 45.9, H 6.6, N 5.7%; C10H16NO5P requires: C 46.0, H 6.2, N 5.4%. Crystals suitable for single-crystal X-ray diffraction were selected directly from the prepared sample.
Space group P21/c was uniquely assigned from the systematic absences. All H atoms were located from difference maps and were thereafter treated as riding atoms, with C—H distances of 0.95 (aromatic), 0.98 (CH3), 0.99 (CH2) and 1.00 Å (aliphatic CH), N—H distances of 0.91 Å, and O—H distances of 0.84 Å.
Data collection: KappaCCD Server Software (Nonius, 1997); cell refinement: DENZO–SMN (Otwinowski & Minor, 1997); data reduction: DENZO–SMN; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: PLATON (Spek, 2003); software used to prepare material for publication: SHELXL97 (Sheldrick, 1997) and PRPKAPPA (Ferguson, 1999).
rac-2-Phenylethylammonium carboxymethylphosphonate(1-)
top
Crystal data top
| C8H12N+·C2H4O5P− | F(000) = 552 |
| Mr = 261.21 | Dx = 1.394 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 2845 reflections |
| a = 8.2969 (3) Å | θ = 2.9–27.5° |
| b = 13.6898 (3) Å | µ = 0.23 mm−1 |
| c = 11.0388 (4) Å | T = 150 K |
| β = 96.7869 (18)° | Plate, colourless |
| V = 1245.03 (7) Å3 | 0.28 × 0.18 × 0.08 mm |
| Z = 4 | |
Data collection top
Nonius KappaCCD
diffractometer | 2394 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed X-ray tube | Rint = 0.039 |
| Graphite monochromator | θmax = 27.5°, θmin = 2.9° |
| ϕ scans, and ω scans with κ offsets | h = −10→10 |
| 10538 measured reflections | k = −15→17 |
| 2845 independent reflections | l = −14→14 |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.036 | H-atom parameters constrained |
| wR(F2) = 0.095 | w = 1/[σ2(Fo2) + (0.0401P)2 + 0.6945P]
where P = (Fo2 + 2Fc2)/3 |
| S = 1.05 | (Δ/σ)max < 0.001 |
| 2845 reflections | Δρmax = 0.30 e Å−3 |
| 159 parameters | Δρmin = −0.33 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0076 (19) |
Crystal data top
| C8H12N+·C2H4O5P− | V = 1245.03 (7) Å3 |
| Mr = 261.21 | Z = 4 |
| Monoclinic, P21/c | Mo Kα radiation |
| a = 8.2969 (3) Å | µ = 0.23 mm−1 |
| b = 13.6898 (3) Å | T = 150 K |
| c = 11.0388 (4) Å | 0.28 × 0.18 × 0.08 mm |
| β = 96.7869 (18)° | |
Data collection top
Nonius KappaCCD
diffractometer | 2394 reflections with I > 2σ(I) |
| 10538 measured reflections | Rint = 0.039 |
| 2845 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.036 | 0 restraints |
| wR(F2) = 0.095 | H-atom parameters constrained |
| S = 1.05 | Δρmax = 0.30 e Å−3 |
| 2845 reflections | Δρmin = −0.33 e Å−3 |
| 159 parameters | |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| P1 | 0.40173 (5) | 0.36759 (3) | 0.44266 (3) | 0.01881 (13) | |
| O1 | 0.29772 (17) | 0.29493 (8) | 0.15716 (11) | 0.0320 (3) | |
| O2 | 0.40129 (15) | 0.44255 (8) | 0.13225 (10) | 0.0278 (3) | |
| O3 | 0.35709 (14) | 0.42341 (8) | 0.55175 (9) | 0.0234 (3) | |
| O4 | 0.39058 (14) | 0.25824 (8) | 0.45235 (9) | 0.0250 (3) | |
| O5 | 0.57737 (13) | 0.39345 (8) | 0.41434 (10) | 0.0233 (3) | |
| C1 | 0.32976 (18) | 0.38536 (11) | 0.19124 (13) | 0.0205 (3) | |
| C2 | 0.2695 (2) | 0.41036 (12) | 0.31071 (14) | 0.0249 (3) | |
| N1 | 0.35240 (15) | 0.09402 (9) | 0.30670 (11) | 0.0202 (3) | |
| C11 | 0.05149 (19) | 0.10557 (11) | 0.29944 (15) | 0.0249 (3) | |
| C12 | −0.0543 (2) | 0.13605 (16) | 0.3801 (2) | 0.0461 (5) | |
| C13 | −0.2009 (3) | 0.18031 (18) | 0.3377 (3) | 0.0663 (8) | |
| C14 | −0.2423 (3) | 0.19524 (15) | 0.2149 (3) | 0.0636 (8) | |
| C15 | −0.1371 (3) | 0.16794 (16) | 0.1346 (2) | 0.0551 (7) | |
| C16 | 0.0095 (2) | 0.12237 (14) | 0.17573 (17) | 0.0351 (4) | |
| C17 | 0.20171 (19) | 0.04885 (11) | 0.34650 (14) | 0.0234 (3) | |
| C18 | 0.1913 (2) | −0.05743 (12) | 0.30635 (19) | 0.0352 (4) | |
| H1 | 0.3347 | 0.2842 | 0.0908 | 0.048* | |
| H5 | 0.5956 | 0.4530 | 0.4284 | 0.035* | |
| H2A | 0.1605 | 0.3814 | 0.3123 | 0.030* | |
| H2B | 0.2583 | 0.4822 | 0.3162 | 0.030* | |
| H1A | 0.3682 | 0.1542 | 0.3412 | 0.030* | |
| H1B | 0.3405 | 0.0998 | 0.2240 | 0.030* | |
| H1C | 0.4395 | 0.0553 | 0.3309 | 0.030* | |
| H12 | −0.0263 | 0.1266 | 0.4653 | 0.055* | |
| H13 | −0.2729 | 0.2003 | 0.3939 | 0.080* | |
| H14 | −0.3435 | 0.2244 | 0.1859 | 0.076* | |
| H15 | −0.1642 | 0.1802 | 0.0500 | 0.066* | |
| H16 | 0.0808 | 0.1028 | 0.1190 | 0.042* | |
| H17 | 0.2124 | 0.0503 | 0.4377 | 0.028* | |
| H18A | 0.1829 | −0.0608 | 0.2172 | 0.053* | |
| H18B | 0.0953 | −0.0877 | 0.3344 | 0.053* | |
| H18C | 0.2889 | −0.0922 | 0.3416 | 0.053* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| P1 | 0.0262 (2) | 0.0155 (2) | 0.0153 (2) | −0.00028 (14) | 0.00470 (14) | −0.00098 (14) |
| O1 | 0.0533 (8) | 0.0220 (6) | 0.0230 (6) | −0.0106 (5) | 0.0142 (5) | −0.0046 (5) |
| O2 | 0.0374 (7) | 0.0238 (6) | 0.0217 (6) | −0.0075 (5) | 0.0010 (5) | 0.0024 (4) |
| O3 | 0.0335 (6) | 0.0200 (5) | 0.0183 (5) | −0.0026 (4) | 0.0091 (4) | −0.0027 (4) |
| O4 | 0.0398 (7) | 0.0161 (5) | 0.0194 (5) | −0.0020 (4) | 0.0051 (5) | −0.0003 (4) |
| O5 | 0.0259 (6) | 0.0214 (5) | 0.0234 (5) | −0.0004 (4) | 0.0064 (4) | −0.0037 (4) |
| C1 | 0.0223 (7) | 0.0202 (7) | 0.0182 (7) | 0.0014 (6) | −0.0014 (6) | 0.0007 (6) |
| C2 | 0.0284 (8) | 0.0248 (8) | 0.0216 (7) | 0.0066 (6) | 0.0037 (6) | −0.0019 (6) |
| N1 | 0.0238 (7) | 0.0173 (6) | 0.0195 (6) | −0.0004 (5) | 0.0026 (5) | −0.0009 (5) |
| C11 | 0.0236 (8) | 0.0192 (7) | 0.0316 (8) | −0.0021 (6) | 0.0022 (6) | −0.0012 (6) |
| C12 | 0.0321 (10) | 0.0553 (13) | 0.0513 (12) | 0.0009 (9) | 0.0063 (9) | −0.0264 (10) |
| C13 | 0.0289 (11) | 0.0528 (14) | 0.117 (2) | 0.0046 (9) | 0.0087 (13) | −0.0476 (16) |
| C14 | 0.0276 (11) | 0.0228 (9) | 0.134 (3) | 0.0009 (8) | −0.0152 (14) | 0.0002 (12) |
| C15 | 0.0353 (11) | 0.0455 (12) | 0.0782 (16) | −0.0146 (9) | −0.0201 (11) | 0.0343 (12) |
| C16 | 0.0278 (9) | 0.0392 (10) | 0.0368 (10) | −0.0087 (7) | −0.0021 (7) | 0.0147 (8) |
| C17 | 0.0255 (8) | 0.0253 (8) | 0.0196 (7) | −0.0019 (6) | 0.0042 (6) | 0.0033 (6) |
| C18 | 0.0308 (9) | 0.0213 (8) | 0.0539 (12) | −0.0016 (7) | 0.0070 (8) | 0.0045 (8) |
Geometric parameters (Å, º) top
| P1—O3 | 1.5088 (11) | C11—C16 | 1.388 (2) |
| P1—O4 | 1.5043 (11) | C11—C17 | 1.508 (2) |
| P1—O5 | 1.5667 (11) | C12—C13 | 1.390 (3) |
| P1—C2 | 1.8137 (16) | C12—H12 | 0.95 |
| C1—O1 | 1.3120 (18) | C13—C14 | 1.374 (4) |
| C1—O2 | 1.2167 (19) | C13—H13 | 0.95 |
| O1—H1 | 0.84 | C14—C15 | 1.367 (4) |
| O5—H5 | 0.84 | C14—H14 | 0.95 |
| C1—C2 | 1.504 (2) | C15—C16 | 1.394 (3) |
| C2—H2A | 0.99 | C15—H15 | 0.95 |
| C2—H2B | 0.99 | C16—H16 | 0.95 |
| N1—C17 | 1.5062 (19) | C17—C18 | 1.520 (2) |
| N1—H1A | 0.91 | C17—H17 | 1.00 |
| N1—H1B | 0.91 | C18—H18A | 0.98 |
| N1—H1C | 0.91 | C18—H18B | 0.98 |
| C11—C12 | 1.386 (3) | C18—H18C | 0.98 |
| | | |
| O3—P1—O4 | 115.10 (6) | C11—C12—H12 | 119.7 |
| O4—P1—O5 | 107.79 (6) | C13—C12—H12 | 119.7 |
| O5—P1—O3 | 111.51 (6) | C14—C13—C12 | 120.2 (2) |
| O3—P1—C2 | 107.18 (7) | C14—C13—H13 | 119.9 |
| O4—P1—C2 | 109.99 (7) | C12—C13—H13 | 119.9 |
| O5—P1—C2 | 104.79 (7) | C15—C14—C13 | 119.6 (2) |
| C1—O1—H1 | 109.5 | C15—C14—H14 | 120.2 |
| P1—O5—H5 | 109.5 | C13—C14—H14 | 120.2 |
| O2—C1—O1 | 123.29 (14) | C14—C15—C16 | 120.7 (2) |
| O2—C1—C2 | 123.81 (14) | C14—C15—H15 | 119.6 |
| O1—C1—C2 | 112.90 (13) | C16—C15—H15 | 119.6 |
| C1—C2—P1 | 113.50 (11) | C11—C16—C15 | 120.1 (2) |
| C1—C2—H2A | 108.9 | C11—C16—H16 | 119.9 |
| P1—C2—H2A | 108.9 | C15—C16—H16 | 119.9 |
| C1—C2—H2B | 108.9 | N1—C17—C11 | 111.65 (12) |
| P1—C2—H2B | 108.9 | N1—C17—C18 | 109.18 (13) |
| H2A—C2—H2B | 107.7 | C11—C17—C18 | 112.09 (14) |
| C17—N1—H1A | 109.5 | N1—C17—H17 | 107.9 |
| C17—N1—H1B | 109.5 | C11—C17—H17 | 107.9 |
| H1A—N1—H1B | 109.5 | C18—C17—H17 | 107.9 |
| C17—N1—H1C | 109.5 | C17—C18—H18A | 109.5 |
| H1A—N1—H1C | 109.5 | C17—C18—H18B | 109.5 |
| H1B—N1—H1C | 109.5 | H18A—C18—H18B | 109.5 |
| C12—C11—C16 | 118.55 (18) | C17—C18—H18C | 109.5 |
| C12—C11—C17 | 119.59 (17) | H18A—C18—H18C | 109.5 |
| C16—C11—C17 | 121.69 (16) | H18B—C18—H18C | 109.5 |
| C11—C12—C13 | 120.7 (2) | | |
| | | |
| O2—C1—C2—P1 | −99.72 (16) | C13—C14—C15—C16 | 2.0 (3) |
| O1—C1—C2—P1 | 80.00 (16) | C12—C11—C16—C15 | −0.7 (3) |
| O4—P1—C2—C1 | −71.68 (13) | C17—C11—C16—C15 | 174.61 (16) |
| O3—P1—C2—C1 | 162.51 (11) | C14—C15—C16—C11 | −1.0 (3) |
| O5—P1—C2—C1 | 43.93 (13) | C12—C11—C17—N1 | −127.50 (16) |
| C16—C11—C12—C13 | 1.5 (3) | C16—C11—C17—N1 | 57.2 (2) |
| C17—C11—C12—C13 | −173.91 (19) | C12—C11—C17—C18 | 109.65 (18) |
| C11—C12—C13—C14 | −0.6 (3) | C16—C11—C17—C18 | −65.7 (2) |
| C12—C13—C14—C15 | −1.2 (3) | | |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O4i | 0.84 | 1.75 | 2.5781 (15) | 169 |
| O5—H5···O3ii | 0.84 | 1.75 | 2.5834 (15) | 176 |
| N1—H1A···O4 | 0.91 | 1.88 | 2.7603 (16) | 164 |
| N1—H1B···O3i | 0.91 | 1.95 | 2.8293 (16) | 162 |
| N1—H1C···O2iii | 0.91 | 2.04 | 2.9334 (17) | 166 |
| C2—H2B···Cg1iv | 0.99 | 2.75 | 3.6417 (19) | 150 |
| Symmetry codes: (i) x, −y+1/2, z−1/2; (ii) −x+1, −y+1, −z+1; (iii) −x+1, y−1/2, −z+1/2; (iv) −x, y+1/2, −z+1/2. |
Experimental details
| Crystal data |
| Chemical formula | C8H12N+·C2H4O5P− |
| Mr | 261.21 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 150 |
| a, b, c (Å) | 8.2969 (3), 13.6898 (3), 11.0388 (4) |
| β (°) | 96.7869 (18) |
| V (Å3) | 1245.03 (7) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.23 |
| Crystal size (mm) | 0.28 × 0.18 × 0.08 |
| |
| Data collection |
| Diffractometer | Nonius KappaCCD
diffractometer |
| Absorption correction | – |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 10538, 2845, 2394 |
| Rint | 0.039 |
| (sin θ/λ)max (Å−1) | 0.650 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.036, 0.095, 1.05 |
| No. of reflections | 2845 |
| No. of parameters | 159 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.30, −0.33 |
Selected geometric parameters (Å, º) top| P1—O3 | 1.5088 (11) | P1—C2 | 1.8137 (16) |
| P1—O4 | 1.5043 (11) | C1—O1 | 1.3120 (18) |
| P1—O5 | 1.5667 (11) | C1—O2 | 1.2167 (19) |
| | | |
| O3—P1—O4 | 115.10 (6) | O3—P1—C2 | 107.18 (7) |
| O4—P1—O5 | 107.79 (6) | O4—P1—C2 | 109.99 (7) |
| O5—P1—O3 | 111.51 (6) | O5—P1—C2 | 104.79 (7) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O4i | 0.84 | 1.75 | 2.5781 (15) | 169 |
| O5—H5···O3ii | 0.84 | 1.75 | 2.5834 (15) | 176 |
| N1—H1A···O4 | 0.91 | 1.88 | 2.7603 (16) | 164 |
| N1—H1B···O3i | 0.91 | 1.95 | 2.8293 (16) | 162 |
| N1—H1C···O2iii | 0.91 | 2.04 | 2.9334 (17) | 166 |
| C2—H2B···Cg1iv | 0.99 | 2.75 | 3.6417 (19) | 150 |
| Symmetry codes: (i) x, −y+1/2, z−1/2; (ii) −x+1, −y+1, −z+1; (iii) −x+1, y−1/2, −z+1/2; (iv) −x, y+1/2, −z+1/2. |


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2004/03/00/sk1696/sk1696fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2004/03/00/sk1696/sk1696fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2004/03/00/sk1696/sk1696fig3thm.gif)




The structures of carboxymethylphosphonic acid [HOOCCH2P(O)(OH)2] and its ammonium salt [NH4]+·[C2H4O5P]− were reported several years ago (Lis, 1997), and since then the supramolecular structures have been reported for a number of salts formed by reaction of this acid with a range of organic diamines (Farrell et al., 2001; Videnova-Adrabinska, 2002; Bowes et al., 2003). We report here the supramolecular structure of the salt, (I), formed with a racemic chiral mono-amine, 2-phenylethylamine.
Compound (I) is a salt, [PhCH(CH3)NH3]+·[HOOCCH2P(O)2OH]− (Fig. 1), in which one H atom has been fully transferred to the amine from a POH unit of the acid; in the anion the C—O and P—O distances (Table 1) are fully consistent with the locations of the associated H atoms as deduced from difference maps. The O—P—O angle involving the two unprotonated O atoms (O3 and O4) is significantly larger than the other O—P—O angles, and it is balanced by the correspondingly small O5—P1—C2 angle. The remaining bond distances and interbond angles show no unusual features. The centrosymmetric space group accommodates equal numbers of (R) and (S) cations, and the asymmetric unit was selected to include a cation of (R) configuration.
The two-dimensional anion substructure is readily analysed in terms of the actions of the two independent O—H···O hydrogen bonds (Table 2), and it is convenient to consider these in turn. Phosphonic atom O5 in the anion at (x, y, z) acts as a hydrogen-bond donor to phosphonate atom O3 in the anion at (1 − x, 1 − y, 1 − z), so generating a centrosymmetric R22(8) ring, centred at (1/2, 1/2, 1/2). The action of the second O—H···O hydrogen bond is then readily analysed in terms of the linking of the R22(8) dimers. Carboxy atoms O1 in the anions at (x, y, z) and (1 − x, 1 − y, 1 − z), which form the (1/2, 1/2, 1/2) dimer, act as hydrogen-bond donors to phosphonate atoms O4 at (x, 0.5 − y, −0.5 + z) and (1 − x, 0.5 + y, 1.5 − z), respectively, which are themselves components of the dimers centred at (1/2, 0, 0) and (1/2, 1, 10), respectively. Similarly, the two O4 atoms in the (1/2, 1/2, 1/2) dimer accept hydrogen bonds from carboxy atoms O1 in the anions at (x, 0.5 − y, 0.5 + z) and (1 − x, 0.5 + y, 0.5 − z), themselves components of the dimers centred at (1/2, 0, 1) and (1/2, 1, 0), respectively.
In this way, the two O—H···O hydrogen bonds generate a (100) sheet of anions built from alternating R22(8) and R66(32) rings, where both ring types are centrosymmetric (Fig. 2). The R22(8) rings are centred at (1/2, m, n) and (1/2, m + 1/2, n + 1/2), and the R66(32) rings are centred at (1/2, m, n + 1/2) and (1/2, m + 1/2, n) (where m and n independently take the value zero or integer). A single sheet of this type passes through each unit cell. The two-dimensional anion substructure of (I) is thus the same as that observed in the imidazolium salt (Videnova-Adrabinska, 2002) but differs from that found in the salt formed with 1,2-bis(4'-pyridyl)ethane, where the anion sheets consist of alternating R22(12) and R66(28) rings (Bowes et al., 2003). A more common occurrence of the R22(8) motif in salts with diamines is as a component of one-dimensional anion substructures including chains of spiro-fused rings and chains of edge-fused rings (Bowes et al., 2003).
The cation in (I) is linked to the anion sheet in a multi-point interaction; the acceptors in the three N—H···O hydrogen bonds are provided to three different anions, all lying in the same (100) sheet (Table 2). Because the anion sheet is centrosymmetric, all of the cations pendent from one face of the sheet have the (R) configuration, while all those pendent from the opposite face of the same sheet have the (S) configuration. Two cations, one (R) and one (S), lie over each R66(32) ring and their effect is to divide this ring into five sectors, viz. one centrosymmetric R46(16) ring and a pair each of R23(10) and R33(12) rings (Fig. 2). The (100) sheet is thus tripartite in nature, with a polar central layer between lipophilic outer layers comprising methyl and phenyl groups (Fig. 3). A rather similar overall structure was observed in the imidazolium salt, although with only two-point attachment of the cations to the anion sheet.
Adjacent (100) sheets are linked weakly by a single C—H···π(arene) hydrogen bond (Table 2); atom C2 in the anion at (x, y, z), part of the sheet centred at x = 1/2, acts as a hydrogen-bond donor, via atom H2B, to the phenyl ring of the cation at (-x, 0.5 + y, 0.5 − z), which forms part of the sheet centred at x = −0.5. Propagation by inversion of this interaction links all of the (100) sheets into a single three-dimensional framework structure.
All of the hard (Desiraju & Steiner, 1999) hydrogen bonds in (I) have D—H···A units that are almost linear and the O—H···O hydrogen bonds are short for their type. It remains a moot point whether the formation of the anion substructure is controlled primarily by the short strong O—H···O hydrogen bonds, or whether the cations provide some significant direction to the structure-assembly process; the formation of the same anion substructure in the presence of two very different cations, imidazolium and 2-phenylethylammonium, perhaps argues against the cation-template effect previously suggested (Videnova-Adrabinska, 2002).