Molecules of the title compound, C
7H
7NO
3S, are linked into centrosymmetric
R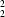
(10) dimers by paired C-H

O hydrogen bonds, and these dimers are linked into [110] chains by a single aromatic
![[pi]](/logos/entities/pi_rmgif.gif)
-
stacking interaction.
Supporting information
CCDC reference: 231088
A sample of compound (I) was prepared by oxidation of a commercial sample of methyl 4-nitrophenyl sulfide (Aldrich), using hydrogen peroxide and TiCl3, following the published procedure of Watanabe et al. (1981). Crystals of (I) suitable for single-crystal X-ray diffraction were grown by slow evaporation of a solution in ethanol (m.p. 430–432 K).
Crystals of (I) are triclinic. Space group P-1 was selected and confirmed by the successful structure analysis. All H atoms were located from difference maps and subsequently treated as riding atoms, with C—H distances 0.95 (aromatic) and 0.98 Å (methyl).
Data collection: KappaCCD Server Software (Nonius, 1997); cell refinement: DENZO-SMN (Otwinowski & Minor, 1997); data reduction: DENZO-SMN; program(s) used to solve structure: OSCAIL (McArdle, 2003) and SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: OSCAIL and SHELXL97 (Sheldrick, 1997; molecular graphics: PLATON (Spek, 2003); software used to prepare material for publication: SHELXL97 (Sheldrick, 1997) and PRPKAPPA (Ferguson, 1999).
Methyl 4-nitrophenyl sulfoxide
top
Crystal data top
| C7H7NO3S | Z = 2 |
| Mr = 185.20 | F(000) = 192 |
| Triclinic, P1 | Dx = 1.603 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation, λ = 0.71073 Å |
| a = 6.1522 (2) Å | Cell parameters from 1739 reflections |
| b = 7.8550 (3) Å | θ = 3.3–27.5° |
| c = 7.9939 (3) Å | µ = 0.38 mm−1 |
| α = 83.684 (3)° | T = 120 K |
| β = 89.320 (2)° | Block, colourless |
| γ = 88.430 (3)° | 0.35 × 0.35 × 0.30 mm |
| V = 383.81 (2) Å3 | |
Data collection top
Nonius KappaCCD area-detector
diffractometer | 1739 independent reflections |
| Radiation source: rotating anode | 1588 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.038 |
| ϕ scans, and ω scans with κ offsets | θmax = 27.5°, θmin = 3.3° |
Absorption correction: multi-scan
(SORTAV; Blessing, 1995, 1997) | h = −7→8 |
| Tmin = 0.878, Tmax = 0.894 | k = −10→9 |
| 6313 measured reflections | l = −10→10 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.030 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.079 | H-atom parameters constrained |
| S = 1.11 | w = 1/[σ2(Fo2) + (0.033P)2 + 0.1817P]
where P = (Fo2 + 2Fc2)/3 |
| 1739 reflections | (Δ/σ)max = 0.001 |
| 110 parameters | Δρmax = 0.32 e Å−3 |
| 0 restraints | Δρmin = −0.41 e Å−3 |
Crystal data top
| C7H7NO3S | γ = 88.430 (3)° |
| Mr = 185.20 | V = 383.81 (2) Å3 |
| Triclinic, P1 | Z = 2 |
| a = 6.1522 (2) Å | Mo Kα radiation |
| b = 7.8550 (3) Å | µ = 0.38 mm−1 |
| c = 7.9939 (3) Å | T = 120 K |
| α = 83.684 (3)° | 0.35 × 0.35 × 0.30 mm |
| β = 89.320 (2)° | |
Data collection top
Nonius KappaCCD area-detector
diffractometer | 1739 independent reflections |
Absorption correction: multi-scan
(SORTAV; Blessing, 1995, 1997) | 1588 reflections with I > 2σ(I) |
| Tmin = 0.878, Tmax = 0.894 | Rint = 0.038 |
| 6313 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.030 | 0 restraints |
| wR(F2) = 0.079 | H-atom parameters constrained |
| S = 1.11 | Δρmax = 0.32 e Å−3 |
| 1739 reflections | Δρmin = −0.41 e Å−3 |
| 110 parameters | |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| C1 | 0.9138 (2) | 0.70819 (17) | 0.43727 (17) | 0.0137 (3) | |
| C2 | 1.1127 (2) | 0.78276 (17) | 0.40236 (17) | 0.0155 (3) | |
| C3 | 1.2219 (2) | 0.84396 (17) | 0.53340 (18) | 0.0152 (3) | |
| C4 | 1.1294 (2) | 0.82927 (17) | 0.69301 (17) | 0.0139 (3) | |
| C5 | 0.9277 (2) | 0.75597 (18) | 0.72548 (17) | 0.0166 (3) | |
| C6 | 0.8181 (2) | 0.69342 (18) | 0.59590 (17) | 0.0156 (3) | |
| N1 | 0.79667 (19) | 0.64214 (15) | 0.29973 (15) | 0.0155 (2) | |
| O1 | 0.87916 (18) | 0.65772 (15) | 0.15837 (13) | 0.0230 (3) | |
| O2 | 0.62245 (17) | 0.57405 (14) | 0.33273 (13) | 0.0226 (2) | |
| S4 | 1.26776 (5) | 0.91596 (4) | 0.86053 (4) | 0.01619 (12) | |
| O41 | 1.45691 (18) | 1.01004 (14) | 0.78095 (13) | 0.0226 (2) | |
| C41 | 1.3715 (2) | 0.71571 (19) | 0.96114 (18) | 0.0184 (3) | |
| H2 | 1.1728 | 0.7918 | 0.2918 | 0.019* | |
| H3 | 1.3591 | 0.8955 | 0.5138 | 0.018* | |
| H5 | 0.8659 | 0.7490 | 0.8354 | 0.020* | |
| H6 | 0.6809 | 0.6417 | 0.6151 | 0.019* | |
| H41A | 1.4571 | 0.6566 | 0.8798 | 0.028* | |
| H41B | 1.2501 | 0.6444 | 1.0033 | 0.028* | |
| H41C | 1.4641 | 0.7366 | 1.0553 | 0.028* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| C1 | 0.0144 (7) | 0.0121 (6) | 0.0143 (6) | 0.0003 (5) | −0.0036 (5) | −0.0005 (5) |
| C2 | 0.0159 (7) | 0.0163 (7) | 0.0138 (6) | −0.0004 (5) | 0.0021 (5) | 0.0001 (5) |
| C3 | 0.0121 (6) | 0.0148 (7) | 0.0186 (7) | −0.0020 (5) | 0.0004 (5) | −0.0004 (5) |
| C4 | 0.0143 (7) | 0.0122 (6) | 0.0152 (6) | 0.0000 (5) | −0.0031 (5) | −0.0020 (5) |
| C5 | 0.0152 (7) | 0.0206 (7) | 0.0139 (6) | −0.0021 (5) | 0.0010 (5) | −0.0006 (5) |
| C6 | 0.0120 (6) | 0.0186 (7) | 0.0159 (7) | −0.0034 (5) | −0.0008 (5) | −0.0001 (5) |
| N1 | 0.0168 (6) | 0.0141 (6) | 0.0154 (6) | −0.0006 (4) | −0.0026 (4) | −0.0011 (4) |
| O1 | 0.0236 (6) | 0.0334 (6) | 0.0124 (5) | −0.0031 (5) | 0.0002 (4) | −0.0045 (4) |
| O2 | 0.0215 (6) | 0.0260 (6) | 0.0208 (5) | −0.0113 (4) | −0.0032 (4) | −0.0020 (4) |
| S4 | 0.0161 (2) | 0.01682 (19) | 0.01650 (19) | −0.00319 (13) | −0.00132 (13) | −0.00508 (13) |
| O41 | 0.0238 (6) | 0.0236 (6) | 0.0211 (5) | −0.0123 (4) | −0.0001 (4) | −0.0031 (4) |
| C41 | 0.0187 (7) | 0.0209 (7) | 0.0159 (7) | −0.0015 (5) | −0.0040 (5) | −0.0030 (5) |
Geometric parameters (Å, º) top
| C1—C2 | 1.3835 (19) | C5—H5 | 0.95 |
| C1—C6 | 1.3863 (19) | C6—H6 | 0.95 |
| C1—N1 | 1.4704 (17) | N1—O2 | 1.2240 (16) |
| C2—C3 | 1.387 (2) | N1—O1 | 1.2283 (16) |
| C2—H2 | 0.95 | S4—O41 | 1.4937 (11) |
| C3—C4 | 1.385 (2) | S4—C41 | 1.7909 (15) |
| C3—H3 | 0.95 | C41—H41A | 0.98 |
| C4—C5 | 1.392 (2) | C41—H41B | 0.98 |
| C4—S4 | 1.8003 (14) | C41—H41C | 0.98 |
| C5—C6 | 1.3841 (19) | | |
| | | |
| C2—C1—C6 | 123.20 (13) | C5—C6—C1 | 118.23 (13) |
| C2—C1—N1 | 118.68 (12) | C5—C6—H6 | 120.9 |
| C6—C1—N1 | 118.11 (12) | C1—C6—H6 | 120.9 |
| C1—C2—C3 | 118.10 (13) | O2—N1—O1 | 123.63 (12) |
| C1—C2—H2 | 120.9 | O2—N1—C1 | 118.07 (11) |
| C3—C2—H2 | 120.9 | O1—N1—C1 | 118.30 (12) |
| C4—C3—C2 | 119.49 (13) | C4—S4—C41 | 96.61 (6) |
| C4—C3—H3 | 120.3 | C4—S4—O41 | 106.29 (6) |
| C2—C3—H3 | 120.3 | O41—S4—C41 | 107.10 (7) |
| C3—C4—C5 | 121.67 (13) | S4—C41—H41A | 109.5 |
| C3—C4—S4 | 119.14 (11) | S4—C41—H41B | 109.5 |
| C5—C4—S4 | 119.13 (10) | H41A—C41—H41B | 109.5 |
| C6—C5—C4 | 119.30 (13) | S4—C41—H41C | 109.5 |
| C6—C5—H5 | 120.4 | H41A—C41—H41C | 109.5 |
| C4—C5—H5 | 120.4 | H41B—C41—H41C | 109.5 |
| | | |
| C6—C1—C2—C3 | 0.7 (2) | N1—C1—C6—C5 | −179.80 (12) |
| N1—C1—C2—C3 | −179.75 (11) | C3—C4—S4—O41 | 6.56 (13) |
| C1—C2—C3—C4 | −0.2 (2) | C5—C4—S4—O41 | −170.63 (11) |
| C2—C3—C4—C5 | −0.7 (2) | C3—C4—S4—C41 | −103.45 (12) |
| C2—C3—C4—S4 | −177.79 (10) | C5—C4—S4—C41 | 79.36 (12) |
| C3—C4—C5—C6 | 1.1 (2) | C2—C1—N1—O1 | −0.87 (19) |
| S4—C4—C5—C6 | 178.24 (10) | C2—C1—N1—O2 | 178.88 (12) |
| C4—C5—C6—C1 | −0.7 (2) | C6—C1—N1—O2 | −1.51 (19) |
| C2—C1—C6—C5 | −0.2 (2) | C6—C1—N1—O1 | 178.74 (12) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C3—H3···O41 | 0.95 | 2.49 | 2.907 (2) | 106 |
| C6—H6···O2i | 0.95 | 2.56 | 3.478 (2) | 164 |
| Symmetry code: (i) −x+1, −y+1, −z+1. |
Experimental details
| Crystal data |
| Chemical formula | C7H7NO3S |
| Mr | 185.20 |
| Crystal system, space group | Triclinic, P1 |
| Temperature (K) | 120 |
| a, b, c (Å) | 6.1522 (2), 7.8550 (3), 7.9939 (3) |
| α, β, γ (°) | 83.684 (3), 89.320 (2), 88.430 (3) |
| V (Å3) | 383.81 (2) |
| Z | 2 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.38 |
| Crystal size (mm) | 0.35 × 0.35 × 0.30 |
| |
| Data collection |
| Diffractometer | Nonius KappaCCD area-detector
diffractometer |
| Absorption correction | Multi-scan
(SORTAV; Blessing, 1995, 1997) |
| Tmin, Tmax | 0.878, 0.894 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 6313, 1739, 1588 |
| Rint | 0.038 |
| (sin θ/λ)max (Å−1) | 0.650 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.030, 0.079, 1.11 |
| No. of reflections | 1739 |
| No. of parameters | 110 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.32, −0.41 |
Selected geometric parameters (Å, º) top| C4—S4 | 1.8003 (14) | S4—C41 | 1.7909 (15) |
| S4—O41 | 1.4937 (11) | | |
| | | |
| C2—C1—C6 | 123.20 (13) | C4—S4—O41 | 106.29 (6) |
| C3—C4—C5 | 121.67 (13) | O41—S4—C41 | 107.10 (7) |
| C4—S4—C41 | 96.61 (6) | | |
| | | |
| C3—C4—S4—O41 | 6.56 (13) | C2—C1—N1—O1 | −0.87 (19) |
| C3—C4—S4—C41 | −103.45 (12) | C2—C1—N1—O2 | 178.88 (12) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C3—H3···O41 | 0.95 | 2.49 | 2.907 (2) | 106 |
| C6—H6···O2i | 0.95 | 2.56 | 3.478 (2) | 164 |
| Symmetry code: (i) −x+1, −y+1, −z+1. |


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2004/01/00/sk1686/sk1686fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2004/01/00/sk1686/sk1686fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2004/01/00/sk1686/sk1686fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/c/issues/2004/01/00/sk1686/sk1686fig4thm.gif)




Molecules of the title compound, (I), are chiral, but the centrosymmetric space group accommodates equal numbers of the (R) and (S) enantiomers; the selected reference molecule (Fig. 1) is of the (R) configuration. Within the molecule, the ring C—C distances span the very narrow range 1.384 (2)–1.392 (2) Å. The two independent C—S distances (Table 1) are very similar but with the distance to the aryl C marginally the longer of the two. The reference mean values for Caryl—S(O)- and Csp3—S(O)- distances (Allen et al., 1987) are 1.790 and 1.809 Å, respectively, while the reference mean value for S═O distances in sulfoxides is 1.497 Å, so that the S—O distance in (I) is entirely characteristic. \sch
The internal C—C—C angles at C1 and C4 in (I), ipso to the electron-withdrawing substituents NO2 and S(O)CH3, respectively, are both greater than 120°, as expected (Domenicano & Murray-Rust, 1979). At the pyramidal S atom, the bond angles involving the sulfoxide atom O41 are both significantly larger than the C—S—C angle. The conformation of the S(O)—CH3 fragment, with the sulfonyl O atom almost coplanar with the aryl ring, may be controlled by an intramolecular dipolar attraction involving the positively charged atom H3 and the negatively charged atom O41. The nitro group is essentially coplanar with the aryl group, and a similar conformation was found in the isomeric compound methyl 2-nitrophenyl sulfoxide, (II) (Ianelli et al., 1992).
The molecules of (I) are weakly linked into centrosymmetric dimers by a nearly linear C—H···O hydrogen bond, and these weak dimers are further linked into chains by a single aromatic π–π stacking interaction. Ring atom C6 in the molecule at (x, y, z), which is adjacent to the nitro group and hence has the most polarized C—H bond in the molecule, acts as hydrogen-bond donor to nitro atom O2 in the molecule at (1 − x, 1 − y, 1 − z), so forming an R22(10) dimer (Bernstein et al., 1995) centred at (1/2, 1/2, 1/2) (Table 2, Fig. 2). The aryl rings of the molecules at (x, y, z) and (2 − x, 2 − y, 1 − z), which are components of the R22(10) dimers centred, respectively, at (1/2, 1/2, 1/2) and (3/2, 3/2, 1/2), are parallel, with an interplanar spacing of 3.367 (2) Å. The centroid separation is 3.666 (2) Å, corresponding to a nearly ideal centroid offset of 1.450 (2) Å. Propagation by inversion of this stacking interaction then generates a chain of π-stacked dimers running parallel to the [110] direction (Fig. 3).
The supramolecular structure of (II) was not discussed at all in the original report (Ianelli et al., 1992). However, use of the atom coordinates for (II) retrieved from the Cambridge Structural Database (CSD; Allen, 2002), CSD refcode KONKON, shows that, in contrast with the supramolecular structure of (I), that of (II) consists of isolated C(7) zigzag chains built from a single C—H···O hydrogen bond and generated by a glide plane in space group P21/c (Fig. 4). π–π stacking interactions are absent from the structure of (II).