

 journal menu
journal menu research papers
research papers









A two-body interatomic potential function, including fractional atomic charges and a shell model for oxygen, and supplemented by an O—Al—O bond-angle energy term, was fitted to the structural, elastic and vibrational properties of  -Al2O3, corundum, at ambient conditions. Full quasi-harmonic calculations were then carried out on a p,T grid of 54 points in the domain 0–40 GPa and 300–1700 K. The crystal structure was equilibrated at each point, taking into account the anisotropy of vibrational pressure and the thermal dependence of elastic constants, so as to obtain unit-cell edges, atomic coordinates, bulk modulus, thermal expansion coefficient and other thermodynamic properties. Polynomial approximations were developed to represent the p,T dependence of these quantities. Comparison with experimental results for the separate p (T = 300 K) and T (p = 0) behaviours shows very good agreement, with average deviations of 0.1% for the unit-cell volume and 6% for the thermal expansion coefficient. The coupled p,T dependence of the properties of corundum is predicted to be very small for the bulk modulus
-Al2O3, corundum, at ambient conditions. Full quasi-harmonic calculations were then carried out on a p,T grid of 54 points in the domain 0–40 GPa and 300–1700 K. The crystal structure was equilibrated at each point, taking into account the anisotropy of vibrational pressure and the thermal dependence of elastic constants, so as to obtain unit-cell edges, atomic coordinates, bulk modulus, thermal expansion coefficient and other thermodynamic properties. Polynomial approximations were developed to represent the p,T dependence of these quantities. Comparison with experimental results for the separate p (T = 300 K) and T (p = 0) behaviours shows very good agreement, with average deviations of 0.1% for the unit-cell volume and 6% for the thermal expansion coefficient. The coupled p,T dependence of the properties of corundum is predicted to be very small for the bulk modulus 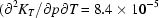 K−1), but not at all negligible for the volume [(1/V)
K−1), but not at all negligible for the volume [(1/V)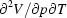 in the range −1.2 to −7.5 × 10−7 GPa−1 K−1 over the p,T domain explored].
in the range −1.2 to −7.5 × 10−7 GPa−1 K−1 over the p,T domain explored].




