Furan-2,5-dicarboxylic acid (FDCA) has been ranked among the top 12 bio-based building-block chemicals by the Department of Energy in the US. The molecule was first synthesized in 1876, but large-scale production has only become possible since the development of modern bio- and chemical catalysis techniques. The structures of two FDCA solvates, namely, FDCA dimethylformamide (DMF) disolvate, C
6H
4O
5·2C
3H
7NO, (I), and FDCA dimethyl sulfoxide (DMSO) monosolvate, C
6H
4O
5·C
2H
6OS, (II), are reported. Solvate (I) crystallizes in the orthorhombic
Pbcn space group and solvate (II) crystallizes in the triclinic
P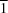
space group. In (I), hydrogen bonds form between the carbonyl O atom in DMF and a hydroxy H atom in FDCA. Whilst in (II), the O atom in one DMSO molecule hydrogen bonds with hydroxy H atoms in two FDCA molecules. Combined with intermolecular S

O interactions, FDCA molecules form a two-dimensional network coordinated by DMSO.
Supporting information
CCDC references: 1857085; 1857084
For both structures, data collection: APEX2 (Bruker, 2010). Cell refinement: APEX2 (Bruker, 2010) for (I); APEX2 and SAINT (Bruker, 2010) for (II). For both structures, data reduction: APEX2 and SAINT (Bruker, 2010); program(s) used to solve structure: SHELXS2015 (Sheldrick, 2015a). Program(s) used to refine structure: SHELXL2017 (Sheldrick, 2015b) for (I); SHELXL2015 (Sheldrick, 2015b) for (II). For both structures, molecular graphics: OLEX2 (Dolomanov et al., 2009), Mercury (Macrae et al., 2008) and Marvin (ChemAxon, 2018); software used to prepare material for publication: APEX2 (Bruker, 2010), SHELXL2015 (Sheldrick, 2015b), PLATON (Spek, 2009) and publCIF (Westrip, 2010).
Furan-2,5-dicarboxylic acid dimethylformamide disolvate (I)
top
Crystal data top
| C6H4O5·2C3H7NO | Dx = 1.324 Mg m−3 |
| Mr = 302.28 | Mo Kα radiation, λ = 0.71073 Å |
| Orthorhombic, Pbcn | Cell parameters from 3053 reflections |
| a = 7.5583 (7) Å | θ = 2.6–23.5° |
| b = 12.7016 (11) Å | µ = 0.11 mm−1 |
| c = 15.7903 (14) Å | T = 150 K |
| V = 1515.9 (2) Å3 | Prism, colourless |
| Z = 4 | 0.19 × 0.15 × 0.05 mm |
| F(000) = 640 | |
Data collection top
Bruker SMART APEXII CCD
diffractometer | 1574 independent reflections |
| Radiation source: sealed tube | 1242 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.032 |
| Detector resolution: 8.333 pixels mm-1 | θmax = 26.5°, θmin = 2.6° |
| φ and ω scans | h = −9→9 |
Absorption correction: multi-scan
(SADABS; Sheldrick, 2008) | k = −15→15 |
| Tmin = 0.905, Tmax = 0.995 | l = −19→19 |
| 15185 measured reflections | |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.030 | All H-atom parameters refined |
| wR(F2) = 0.067 | w = 1/[σ2(Fo2) + (0.006P)2 + 0.632P]
where P = (Fo2 + 2Fc2)/3 |
| S = 1.00 | (Δ/σ)max < 0.001 |
| 1574 reflections | Δρmax = 0.17 e Å−3 |
| 133 parameters | Δρmin = −0.15 e Å−3 |
| 0 restraints | Extinction correction: SHELXL2017 (Sheldrick, 2015b), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0025 (6) |
Special details top
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell esds are taken
into account individually in the estimation of esds in distances, angles
and torsion angles; correlations between esds in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| O1 | 1.000000 | 0.58236 (8) | 0.250000 | 0.0300 (3) | |
| H1 | 0.8481 (17) | 0.3572 (10) | 0.2881 (8) | 0.035 (4)* | |
| C1 | 0.91710 (17) | 0.41487 (9) | 0.27058 (8) | 0.0307 (3) | |
| C2 | 0.87185 (16) | 0.51732 (9) | 0.28191 (7) | 0.0286 (3) | |
| C3 | 0.71524 (17) | 0.56764 (9) | 0.31968 (8) | 0.0324 (3) | |
| O2 | 0.69092 (12) | 0.66197 (7) | 0.32117 (6) | 0.0423 (3) | |
| O3 | 0.60801 (14) | 0.49647 (7) | 0.35065 (7) | 0.0480 (3) | |
| H3 | 0.504 (3) | 0.5251 (15) | 0.3738 (12) | 0.093 (7)* | |
| O4 | 0.33028 (12) | 0.56295 (7) | 0.42123 (6) | 0.0439 (3) | |
| C4 | 0.26531 (18) | 0.64791 (10) | 0.39776 (9) | 0.0347 (3) | |
| H4 | 0.3123 (18) | 0.6863 (11) | 0.3475 (9) | 0.044 (4)* | |
| N5 | 0.13253 (14) | 0.69539 (8) | 0.43592 (7) | 0.0343 (3) | |
| C6 | 0.0592 (2) | 0.79398 (12) | 0.40392 (12) | 0.0485 (4) | |
| H61 | 0.126 (2) | 0.8152 (13) | 0.3542 (11) | 0.068 (5)* | |
| H62 | −0.066 (3) | 0.7834 (13) | 0.3884 (10) | 0.068 (5)* | |
| H63 | 0.068 (2) | 0.8482 (14) | 0.4489 (11) | 0.065 (5)* | |
| C7 | 0.0510 (2) | 0.65109 (14) | 0.51110 (11) | 0.0489 (4) | |
| H71 | 0.117 (2) | 0.5881 (14) | 0.5279 (10) | 0.064 (5)* | |
| H72 | 0.052 (2) | 0.7064 (14) | 0.5575 (11) | 0.076 (6)* | |
| H73 | −0.072 (2) | 0.6331 (13) | 0.4989 (10) | 0.067 (5)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| O1 | 0.0336 (6) | 0.0223 (6) | 0.0341 (7) | 0.000 | 0.0021 (5) | 0.000 |
| C1 | 0.0376 (7) | 0.0233 (6) | 0.0311 (7) | −0.0019 (5) | 0.0036 (5) | 0.0013 (5) |
| C2 | 0.0338 (6) | 0.0241 (6) | 0.0278 (6) | −0.0023 (5) | 0.0007 (5) | 0.0024 (5) |
| C3 | 0.0360 (7) | 0.0277 (7) | 0.0334 (7) | 0.0019 (5) | 0.0009 (5) | 0.0018 (5) |
| O2 | 0.0448 (5) | 0.0251 (5) | 0.0570 (6) | 0.0065 (4) | 0.0081 (5) | 0.0021 (4) |
| O3 | 0.0456 (6) | 0.0288 (5) | 0.0697 (7) | 0.0025 (4) | 0.0260 (5) | 0.0042 (5) |
| O4 | 0.0433 (5) | 0.0352 (5) | 0.0531 (6) | 0.0089 (4) | 0.0116 (5) | 0.0008 (4) |
| C4 | 0.0369 (7) | 0.0330 (7) | 0.0343 (7) | −0.0035 (6) | 0.0004 (6) | −0.0038 (6) |
| N5 | 0.0358 (6) | 0.0313 (6) | 0.0359 (6) | 0.0039 (5) | −0.0012 (5) | −0.0003 (5) |
| C6 | 0.0470 (9) | 0.0383 (8) | 0.0601 (11) | 0.0076 (7) | −0.0049 (8) | 0.0064 (8) |
| C7 | 0.0494 (10) | 0.0467 (9) | 0.0505 (10) | 0.0124 (8) | 0.0147 (8) | 0.0060 (8) |
Geometric parameters (Å, º) top
| O1—C2 | 1.3691 (13) | C4—N5 | 1.3168 (16) |
| O1—C2i | 1.3692 (13) | C4—H4 | 0.996 (14) |
| C1—C2 | 1.3574 (16) | N5—C7 | 1.4512 (18) |
| C1—C1i | 1.412 (2) | N5—C6 | 1.4597 (18) |
| C1—H1 | 0.941 (13) | C6—H61 | 0.970 (18) |
| C2—C3 | 1.4716 (17) | C6—H62 | 0.985 (18) |
| C3—O2 | 1.2124 (14) | C6—H63 | 0.991 (17) |
| C3—O3 | 1.3088 (15) | C7—H71 | 0.980 (17) |
| O3—H3 | 0.94 (2) | C7—H72 | 1.015 (18) |
| O4—C4 | 1.2422 (16) | C7—H73 | 0.979 (18) |
| | | |
| C2—O1—C2i | 105.77 (12) | C4—N5—C6 | 121.59 (13) |
| C2—C1—C1i | 106.52 (7) | C7—N5—C6 | 117.03 (12) |
| C2—C1—H1 | 124.6 (8) | N5—C6—H61 | 108.8 (10) |
| C1i—C1—H1 | 128.9 (8) | N5—C6—H62 | 109.5 (10) |
| C1—C2—O1 | 110.59 (11) | H61—C6—H62 | 109.4 (14) |
| C1—C2—C3 | 132.26 (11) | N5—C6—H63 | 108.8 (10) |
| O1—C2—C3 | 117.14 (10) | H61—C6—H63 | 110.6 (14) |
| O2—C3—O3 | 125.55 (12) | H62—C6—H63 | 109.8 (14) |
| O2—C3—C2 | 123.99 (11) | N5—C7—H71 | 108.7 (10) |
| O3—C3—C2 | 110.46 (10) | N5—C7—H72 | 108.5 (10) |
| C3—O3—H3 | 113.3 (12) | H71—C7—H72 | 111.4 (14) |
| O4—C4—N5 | 124.24 (13) | N5—C7—H73 | 109.5 (10) |
| O4—C4—H4 | 121.4 (8) | H71—C7—H73 | 110.4 (14) |
| N5—C4—H4 | 114.3 (8) | H72—C7—H73 | 108.3 (14) |
| C4—N5—C7 | 121.37 (12) | | |
| | | |
| C1i—C1—C2—O1 | 0.09 (17) | O1—C2—C3—O2 | 2.86 (18) |
| C1i—C1—C2—C3 | 179.15 (13) | C1—C2—C3—O3 | 3.7 (2) |
| C2i—O1—C2—C1 | −0.04 (7) | O1—C2—C3—O3 | −177.30 (10) |
| C2i—O1—C2—C3 | −179.26 (13) | O4—C4—N5—C7 | 0.7 (2) |
| C1—C2—C3—O2 | −176.15 (14) | O4—C4—N5—C6 | −178.45 (13) |
| Symmetry code: (i) −x+2, y, −z+1/2. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O3—H3···O4 | 0.94 (2) | 1.59 (2) | 2.5223 (13) | 173.2 (19) |
Furan-2,5-dicarboxylic acid dimethyl sulfoxide monosolvate (II)
top
Crystal data top
| C6H4O5·C2H6OS | Z = 2 |
| Mr = 234.22 | F(000) = 244 |
| Triclinic, P1 | Dx = 1.560 Mg m−3 |
| a = 6.3129 (8) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 6.9099 (9) Å | Cell parameters from 2660 reflections |
| c = 13.0901 (17) Å | θ = 3.2–28.2° |
| α = 80.5164 (17)° | µ = 0.33 mm−1 |
| β = 77.3664 (17)° | T = 150 K |
| γ = 63.8260 (16)° | Plate, colourless |
| V = 498.53 (11) Å3 | 0.17 × 0.15 × 0.04 mm |
Data collection top
Bruker SMART APEXII CCD
diffractometer | 2044 independent reflections |
| Radiation source: sealed tube | 1894 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.018 |
| φ and ω scans | θmax = 26.5°, θmin = 1.6° |
Absorption correction: multi-scan
(TWINABS; Krause et al., 2015) | h = −7→7 |
| Tmin = 0.847, Tmax = 0.987 | k = −8→8 |
| 2044 measured reflections | l = 0→16 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.024 | Hydrogen site location: difference Fourier map |
| wR(F2) = 0.051 | All H-atom parameters refined |
| S = 1.00 | w = 1/[σ2(Fo2) + (0.0057P)2 + 0.3718P]
where P = (Fo2 + 2Fc2)/3 |
| 2044 reflections | (Δ/σ)max < 0.001 |
| 177 parameters | Δρmax = 0.30 e Å−3 |
| 0 restraints | Δρmin = −0.26 e Å−3 |
Special details top
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell esds are taken
into account individually in the estimation of esds in distances, angles
and torsion angles; correlations between esds in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell esds is used for estimating esds involving l.s. planes. |
Refinement. Refined as a 2-component twin but 180 deg rotation around 001 axis in
reciprocal space, yielding 0.9098:0.0902 (9) component ratio. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| O1 | 0.08991 (17) | 0.41577 (17) | 0.24448 (8) | 0.0214 (2) | |
| S1 | 0.07863 (6) | 0.45030 (6) | 0.12667 (3) | 0.01795 (9) | |
| C1 | 0.3126 (3) | 0.2149 (3) | 0.07023 (13) | 0.0224 (3) | |
| H1A | 0.460 (3) | 0.189 (3) | 0.0900 (14) | 0.028 (5)* | |
| H1B | 0.318 (3) | 0.244 (3) | −0.0039 (15) | 0.033 (5)* | |
| H1C | 0.270 (3) | 0.098 (3) | 0.0972 (14) | 0.028 (5)* | |
| C2 | 0.1993 (3) | 0.6431 (3) | 0.07744 (14) | 0.0268 (3) | |
| H2A | 0.086 (4) | 0.777 (3) | 0.1041 (16) | 0.042 (6)* | |
| H2B | 0.218 (3) | 0.654 (3) | 0.0013 (15) | 0.030 (5)* | |
| H2C | 0.348 (3) | 0.596 (3) | 0.1017 (14) | 0.030 (5)* | |
| O11 | 0.65361 (18) | 0.56009 (17) | 0.35203 (8) | 0.0218 (2) | |
| H11 | 0.794 (4) | 0.518 (3) | 0.3177 (17) | 0.047 (6)* | |
| O12 | 0.57910 (18) | 0.77573 (17) | 0.20352 (8) | 0.0245 (2) | |
| C11 | 0.5112 (2) | 0.7081 (2) | 0.28978 (11) | 0.0184 (3) | |
| C12 | 0.2568 (2) | 0.7818 (2) | 0.33674 (11) | 0.0180 (3) | |
| O25 | 0.10056 (17) | 0.93437 (16) | 0.27591 (8) | 0.0183 (2) | |
| C13 | 0.1377 (3) | 0.7310 (2) | 0.42851 (12) | 0.0220 (3) | |
| H13 | 0.205 (3) | 0.635 (3) | 0.4811 (14) | 0.025 (4)* | |
| C14 | −0.1092 (3) | 0.8591 (2) | 0.42580 (12) | 0.0220 (3) | |
| H14 | −0.239 (3) | 0.861 (3) | 0.4763 (14) | 0.027 (5)* | |
| C15 | −0.1224 (2) | 0.9787 (2) | 0.33226 (11) | 0.0180 (3) | |
| C16 | −0.3208 (2) | 1.1374 (2) | 0.27925 (11) | 0.0186 (3) | |
| O61 | −0.53223 (18) | 1.17145 (18) | 0.33764 (8) | 0.0228 (2) | |
| H61 | −0.637 (4) | 1.248 (3) | 0.3064 (16) | 0.043 (6)* | |
| O62 | −0.29003 (18) | 1.22351 (17) | 0.19237 (8) | 0.0241 (2) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| O1 | 0.0147 (5) | 0.0284 (6) | 0.0152 (5) | −0.0045 (4) | −0.0019 (4) | −0.0002 (4) |
| S1 | 0.01411 (17) | 0.02032 (17) | 0.01631 (17) | −0.00464 (13) | −0.00323 (13) | 0.00009 (13) |
| C1 | 0.0191 (8) | 0.0211 (8) | 0.0200 (8) | −0.0023 (6) | −0.0023 (6) | −0.0023 (6) |
| C2 | 0.0315 (9) | 0.0278 (9) | 0.0233 (9) | −0.0157 (7) | −0.0031 (7) | −0.0008 (7) |
| O11 | 0.0132 (5) | 0.0268 (6) | 0.0189 (5) | −0.0041 (4) | −0.0026 (4) | 0.0024 (4) |
| O12 | 0.0184 (5) | 0.0249 (5) | 0.0210 (6) | −0.0042 (4) | −0.0006 (4) | 0.0043 (4) |
| C11 | 0.0171 (7) | 0.0178 (7) | 0.0185 (7) | −0.0052 (6) | −0.0040 (5) | −0.0017 (6) |
| C12 | 0.0170 (7) | 0.0176 (7) | 0.0177 (7) | −0.0047 (6) | −0.0058 (5) | −0.0006 (5) |
| O25 | 0.0131 (5) | 0.0209 (5) | 0.0169 (5) | −0.0046 (4) | −0.0024 (4) | 0.0015 (4) |
| C13 | 0.0191 (7) | 0.0251 (8) | 0.0179 (7) | −0.0066 (6) | −0.0045 (6) | 0.0026 (6) |
| C14 | 0.0173 (7) | 0.0278 (8) | 0.0178 (7) | −0.0084 (6) | −0.0009 (6) | −0.0001 (6) |
| C15 | 0.0140 (7) | 0.0212 (7) | 0.0180 (7) | −0.0068 (6) | −0.0008 (5) | −0.0033 (6) |
| C16 | 0.0162 (7) | 0.0199 (7) | 0.0200 (7) | −0.0072 (6) | −0.0040 (5) | −0.0022 (6) |
| O61 | 0.0127 (5) | 0.0288 (6) | 0.0202 (6) | −0.0037 (4) | −0.0032 (4) | 0.0022 (5) |
| O62 | 0.0195 (5) | 0.0298 (6) | 0.0209 (6) | −0.0102 (5) | −0.0052 (4) | 0.0056 (5) |
Geometric parameters (Å, º) top
| O1—S1 | 1.5336 (10) | C11—C12 | 1.472 (2) |
| S1—C2 | 1.7802 (17) | C12—C13 | 1.353 (2) |
| S1—C1 | 1.7811 (15) | C12—O25 | 1.3667 (16) |
| C1—H1A | 0.953 (18) | O25—C15 | 1.3633 (17) |
| C1—H1B | 0.954 (19) | C13—C14 | 1.418 (2) |
| C1—H1C | 0.951 (19) | C13—H13 | 0.908 (17) |
| C2—H2A | 0.95 (2) | C14—C15 | 1.356 (2) |
| C2—H2B | 0.972 (19) | C14—H14 | 0.930 (18) |
| C2—H2C | 0.957 (19) | C15—C16 | 1.4732 (19) |
| O11—C11 | 1.3238 (17) | C16—O62 | 1.2115 (18) |
| O11—H11 | 0.84 (2) | C16—O61 | 1.3256 (17) |
| O12—C11 | 1.2118 (17) | O61—H61 | 0.79 (2) |
| | | |
| O1—S1—C2 | 104.53 (7) | O11—C11—C12 | 112.26 (12) |
| O1—S1—C1 | 105.94 (7) | C13—C12—O25 | 110.77 (12) |
| C2—S1—C1 | 100.26 (8) | C13—C12—C11 | 134.23 (13) |
| S1—C1—H1A | 109.8 (10) | O25—C12—C11 | 115.00 (12) |
| S1—C1—H1B | 105.9 (11) | C15—O25—C12 | 105.88 (11) |
| H1A—C1—H1B | 111.5 (15) | C12—C13—C14 | 106.34 (13) |
| S1—C1—H1C | 106.4 (10) | C12—C13—H13 | 126.0 (11) |
| H1A—C1—H1C | 110.4 (14) | C14—C13—H13 | 127.6 (11) |
| H1B—C1—H1C | 112.6 (15) | C15—C14—C13 | 106.26 (13) |
| S1—C2—H2A | 106.7 (12) | C15—C14—H14 | 125.7 (11) |
| S1—C2—H2B | 107.3 (10) | C13—C14—H14 | 128.1 (11) |
| H2A—C2—H2B | 110.4 (15) | C14—C15—O25 | 110.75 (12) |
| S1—C2—H2C | 108.6 (11) | C14—C15—C16 | 134.29 (13) |
| H2A—C2—H2C | 111.1 (16) | O25—C15—C16 | 114.93 (12) |
| H2B—C2—H2C | 112.4 (15) | O62—C16—O61 | 125.09 (13) |
| C11—O11—H11 | 106.5 (14) | O62—C16—C15 | 123.00 (13) |
| O12—C11—O11 | 124.71 (13) | O61—C16—C15 | 111.90 (12) |
| O12—C11—C12 | 123.03 (13) | C16—O61—H61 | 111.1 (15) |
| | | |
| O12—C11—C12—C13 | 179.01 (16) | C13—C14—C15—O25 | −0.07 (17) |
| O11—C11—C12—C13 | −0.5 (2) | C13—C14—C15—C16 | 178.07 (16) |
| O12—C11—C12—O25 | −0.3 (2) | C12—O25—C15—C14 | 0.07 (16) |
| O11—C11—C12—O25 | −179.78 (12) | C12—O25—C15—C16 | −178.46 (12) |
| C13—C12—O25—C15 | −0.05 (16) | C14—C15—C16—O62 | −178.39 (16) |
| C11—C12—O25—C15 | 179.44 (12) | O25—C15—C16—O62 | −0.3 (2) |
| O25—C12—C13—C14 | 0.00 (17) | C14—C15—C16—O61 | 1.2 (2) |
| C11—C12—C13—C14 | −179.34 (16) | O25—C15—C16—O61 | 179.33 (12) |
| C12—C13—C14—C15 | 0.04 (18) | | |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O11—H11···O1i | 0.84 (2) | 1.78 (2) | 2.6279 (14) | 177 (2) |
| O61—H61···O1ii | 0.79 (2) | 1.87 (2) | 2.6587 (15) | 173 (2) |
| Symmetry codes: (i) x+1, y, z; (ii) x−1, y+1, z. |


 journal menu
journal menu research papers
research papers















