The title compound, [Co
2(H
2PO
4)
4(C
10H
8N
2)
2], is dinuclear, centred on a symmetry centre of the
P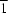
space group. Each Co atom has a distorted square-pyramidal coordination involving two N atoms from a bipyridine molecule and three O atoms from two bridging and one terminal dihydrogen orthophosphate anion. The molecular structure and packing are stabilized by intermolecular hydrogen-bond interactions.
Supporting information
CCDC reference: 245867
The title compound was prepared by the reaction of cobalt dichloride with 2,2'-bipyridine in phosphoric acid solution (Ratios or quantities of starting materials?) by means of hydrothermal synthesis in a stainless-steel reactor with a Teflon liner at 383 K for 24 h.
H atoms were fixed geometrically and allowed to ride on their attached atoms, with C—H distances of 0.93 Å and O—H distances of 0.82 Å, and with Uiso = 1.2Ueq(parent). Please check added text.
Data collection: SMART (Siemens, 1996); cell refinement: SAINT (Siemens, 1996); data reduction: SHELXTL (Sheldrick, 1997a); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997b); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997b); molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL.
bis(µ-dihydrogen phosphato-
κO:
O')bis[(bipyridine-
κ2N,
N')(dihydrogen phosphato-
κO)cobalt(II)]
top
Crystal data top
| [Co(H2PO4)4(C10H8N2)2] | Z = 1 |
| Mr = 818.17 | F(000) = 414 |
| Triclinic, P1 | Dx = 1.857 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation, λ = 0.71073 Å |
| a = 8.6797 (17) Å | Cell parameters from 20 reflections |
| b = 8.7749 (18) Å | θ = 2–11° |
| c = 10.057 (2) Å | µ = 1.44 mm−1 |
| α = 96.09 (3)° | T = 293 K |
| β = 100.55 (3)° | Block, red |
| γ = 100.89 (3)° | 0.20 × 0.20 × 0.10 mm |
| V = 731.6 (3) Å3 | |
Data collection top
Siemens SMART CCD area-detector
diffractometer | 2902 independent reflections |
| Radiation source: fine-focus sealed tube | 2415 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.016 |
| ϕ and ω scans | θmax = 26.2°, θmin = 2.1° |
Absorption correction: empirical (using intensity measurements)
(SADABS; Sheldrick, 1996) | h = −10→10 |
| Tmin = 0.758, Tmax = 0.866 | k = −8→11 |
| 3435 measured reflections | l = −12→12 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.040 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.091 | H-atom parameters constrained |
| S = 1.00 | w = 1/[σ2(Fo2) + (0.0452P)2]
where P = (Fo2 + 2Fc2)/3 |
| 2902 reflections | (Δ/σ)max < 0.001 |
| 208 parameters | Δρmax = 0.41 e Å−3 |
| 0 restraints | Δρmin = −0.32 e Å−3 |
Crystal data top
| [Co(H2PO4)4(C10H8N2)2] | γ = 100.89 (3)° |
| Mr = 818.17 | V = 731.6 (3) Å3 |
| Triclinic, P1 | Z = 1 |
| a = 8.6797 (17) Å | Mo Kα radiation |
| b = 8.7749 (18) Å | µ = 1.44 mm−1 |
| c = 10.057 (2) Å | T = 293 K |
| α = 96.09 (3)° | 0.20 × 0.20 × 0.10 mm |
| β = 100.55 (3)° | |
Data collection top
Siemens SMART CCD area-detector
diffractometer | 2902 independent reflections |
Absorption correction: empirical (using intensity measurements)
(SADABS; Sheldrick, 1996) | 2415 reflections with I > 2σ(I) |
| Tmin = 0.758, Tmax = 0.866 | Rint = 0.016 |
| 3435 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.040 | 0 restraints |
| wR(F2) = 0.091 | H-atom parameters constrained |
| S = 1.00 | Δρmax = 0.41 e Å−3 |
| 2902 reflections | Δρmin = −0.32 e Å−3 |
| 208 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Co1 | 0.05455 (5) | 0.21267 (5) | 0.21455 (4) | 0.02718 (14) | |
| P1 | 0.29540 (10) | 0.43255 (9) | 0.08031 (8) | 0.0313 (2) | |
| P2 | −0.18636 (9) | 0.09242 (9) | −0.07621 (7) | 0.0272 (2) | |
| O1 | 0.2346 (2) | 0.3861 (2) | 0.2034 (2) | 0.0342 (5) | |
| O2 | 0.3964 (3) | 0.5947 (2) | 0.0984 (2) | 0.0391 (6) | |
| O3 | 0.3985 (3) | 0.3126 (3) | 0.0402 (3) | 0.0468 (6) | |
| H3A | 0.4615 | 0.3525 | −0.0044 | 0.070* | |
| O4 | 0.1532 (3) | 0.4170 (3) | −0.0428 (2) | 0.0495 (6) | |
| H4A | 0.0741 | 0.3576 | −0.0298 | 0.074* | |
| O5 | −0.0776 (2) | 0.2252 (2) | 0.02482 (19) | 0.0307 (5) | |
| O6 | −0.2948 (3) | 0.1614 (3) | −0.1843 (2) | 0.0443 (6) | |
| H6A | −0.3205 | 0.2376 | −0.1470 | 0.067* | |
| O7 | −0.2958 (3) | 0.0015 (3) | 0.0081 (2) | 0.0516 (7) | |
| H7A | −0.3151 | −0.0926 | −0.0206 | 0.077* | |
| O8 | −0.1073 (3) | −0.0116 (3) | −0.1538 (2) | 0.0363 (5) | |
| N1 | −0.1037 (3) | 0.2795 (3) | 0.3281 (3) | 0.0347 (6) | |
| N2 | 0.1626 (3) | 0.1908 (3) | 0.4162 (2) | 0.0354 (6) | |
| C1 | −0.2323 (4) | 0.3335 (5) | 0.2773 (4) | 0.0493 (10) | |
| H1A | −0.2580 | 0.3346 | 0.1835 | 0.059* | |
| C2 | −0.3290 (5) | 0.3878 (5) | 0.3577 (4) | 0.0623 (12) | |
| H2B | −0.4192 | 0.4231 | 0.3191 | 0.075* | |
| C3 | −0.2888 (5) | 0.3883 (5) | 0.4956 (4) | 0.0640 (12) | |
| H3B | −0.3501 | 0.4268 | 0.5527 | 0.077* | |
| C4 | −0.1580 (5) | 0.3320 (5) | 0.5493 (4) | 0.0548 (11) | |
| H4B | −0.1308 | 0.3308 | 0.6430 | 0.066* | |
| C5 | −0.0668 (4) | 0.2772 (4) | 0.4643 (3) | 0.0375 (8) | |
| C6 | 0.0784 (4) | 0.2174 (4) | 0.5119 (3) | 0.0375 (8) | |
| C7 | 0.1283 (5) | 0.1898 (5) | 0.6450 (3) | 0.0574 (11) | |
| H7B | 0.0676 | 0.2053 | 0.7104 | 0.069* | |
| C8 | 0.2696 (6) | 0.1390 (5) | 0.6781 (4) | 0.0653 (12) | |
| H8A | 0.3053 | 0.1203 | 0.7666 | 0.078* | |
| C9 | 0.3558 (6) | 0.1165 (5) | 0.5816 (4) | 0.0636 (12) | |
| H9A | 0.4525 | 0.0845 | 0.6031 | 0.076* | |
| C10 | 0.2980 (5) | 0.1416 (5) | 0.4507 (4) | 0.0510 (10) | |
| H10A | 0.3561 | 0.1234 | 0.3838 | 0.061* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Co1 | 0.0312 (2) | 0.0306 (3) | 0.0212 (2) | 0.00929 (18) | 0.00780 (17) | 0.00115 (17) |
| P1 | 0.0341 (5) | 0.0272 (5) | 0.0342 (4) | 0.0056 (4) | 0.0127 (4) | 0.0035 (4) |
| P2 | 0.0307 (4) | 0.0270 (4) | 0.0242 (4) | 0.0096 (3) | 0.0042 (3) | 0.0014 (3) |
| O1 | 0.0343 (12) | 0.0358 (13) | 0.0289 (11) | −0.0016 (10) | 0.0089 (9) | 0.0009 (10) |
| O2 | 0.0379 (13) | 0.0239 (12) | 0.0578 (15) | 0.0044 (10) | 0.0209 (11) | 0.0010 (11) |
| O3 | 0.0532 (15) | 0.0272 (13) | 0.0699 (17) | 0.0091 (11) | 0.0362 (13) | 0.0092 (12) |
| O4 | 0.0477 (15) | 0.0590 (17) | 0.0372 (13) | −0.0017 (12) | 0.0049 (11) | 0.0167 (12) |
| O5 | 0.0359 (12) | 0.0287 (12) | 0.0251 (10) | 0.0079 (9) | 0.0015 (9) | 0.0004 (9) |
| O6 | 0.0564 (15) | 0.0429 (14) | 0.0304 (12) | 0.0266 (12) | −0.0093 (11) | −0.0069 (11) |
| O7 | 0.0644 (17) | 0.0352 (14) | 0.0567 (15) | −0.0017 (12) | 0.0328 (13) | −0.0002 (12) |
| O8 | 0.0435 (13) | 0.0371 (13) | 0.0305 (11) | 0.0190 (11) | 0.0062 (10) | −0.0008 (10) |
| N1 | 0.0364 (15) | 0.0406 (16) | 0.0273 (13) | 0.0067 (13) | 0.0110 (12) | 0.0006 (12) |
| N2 | 0.0399 (16) | 0.0395 (16) | 0.0248 (13) | 0.0098 (13) | 0.0028 (12) | 0.0004 (12) |
| C1 | 0.048 (2) | 0.061 (3) | 0.041 (2) | 0.0222 (19) | 0.0093 (17) | −0.0019 (18) |
| C2 | 0.048 (2) | 0.077 (3) | 0.060 (3) | 0.021 (2) | 0.013 (2) | −0.015 (2) |
| C3 | 0.055 (3) | 0.075 (3) | 0.061 (3) | 0.012 (2) | 0.028 (2) | −0.019 (2) |
| C4 | 0.062 (3) | 0.065 (3) | 0.037 (2) | 0.005 (2) | 0.0236 (19) | −0.0043 (19) |
| C5 | 0.043 (2) | 0.038 (2) | 0.0289 (16) | 0.0008 (16) | 0.0139 (15) | −0.0019 (15) |
| C6 | 0.050 (2) | 0.0349 (19) | 0.0233 (15) | −0.0004 (16) | 0.0080 (15) | 0.0000 (14) |
| C7 | 0.079 (3) | 0.063 (3) | 0.0278 (18) | 0.004 (2) | 0.0144 (19) | 0.0062 (19) |
| C8 | 0.090 (3) | 0.068 (3) | 0.0297 (19) | 0.015 (3) | −0.007 (2) | 0.010 (2) |
| C9 | 0.073 (3) | 0.070 (3) | 0.042 (2) | 0.028 (2) | −0.014 (2) | 0.002 (2) |
| C10 | 0.053 (2) | 0.062 (3) | 0.0370 (19) | 0.022 (2) | 0.0012 (17) | 0.0008 (18) |
Geometric parameters (Å, º) top
| Co1—O8i | 1.968 (2) | N2—C10 | 1.327 (4) |
| Co1—O1 | 1.993 (2) | N2—C6 | 1.337 (4) |
| Co1—O5 | 2.064 (2) | C1—C2 | 1.381 (5) |
| Co1—N1 | 2.067 (3) | C1—H1A | 0.9300 |
| Co1—N2 | 2.117 (3) | C2—C3 | 1.366 (6) |
| P1—O1 | 1.497 (2) | C2—H2B | 0.9300 |
| P1—O2 | 1.497 (2) | C3—C4 | 1.367 (5) |
| P1—O3 | 1.572 (2) | C3—H3B | 0.9300 |
| P1—O4 | 1.554 (2) | C4—C5 | 1.376 (4) |
| P2—O5 | 1.505 (2) | C4—H4B | 0.9300 |
| P2—O6 | 1.554 (2) | C5—C6 | 1.475 (5) |
| P2—O7 | 1.553 (2) | C6—C7 | 1.390 (4) |
| P2—O8 | 1.481 (2) | C7—C8 | 1.379 (6) |
| O3—H3A | 0.8200 | C7—H7B | 0.9300 |
| O4—H4A | 0.8200 | C8—C9 | 1.350 (6) |
| O6—H6A | 0.8200 | C8—H8A | 0.9300 |
| O7—H7A | 0.8200 | C9—C10 | 1.376 (5) |
| O8—P2 | 1.481 (2) | C9—H9A | 0.9300 |
| N1—C1 | 1.332 (4) | C10—H10A | 0.9300 |
| N1—C5 | 1.352 (4) | | |
| | | |
| O8i—Co1—O1 | 108.57 (9) | C10—N2—Co1 | 125.0 (2) |
| O8i—Co1—O5 | 93.73 (9) | C6—N2—Co1 | 115.8 (2) |
| O1—Co1—O5 | 93.94 (9) | N1—C1—C2 | 122.8 (3) |
| O8i—Co1—N1 | 134.82 (11) | N1—C1—H1A | 118.6 |
| O1—Co1—N1 | 114.27 (10) | C2—C1—H1A | 118.6 |
| O5—Co1—N1 | 96.96 (9) | C3—C2—C1 | 118.2 (4) |
| O8i—Co1—N2 | 87.81 (10) | C3—C2—H2B | 120.9 |
| O1—Co1—N2 | 92.14 (10) | C1—C2—H2B | 120.9 |
| O5—Co1—N2 | 172.90 (9) | C2—C3—C4 | 119.7 (3) |
| N1—Co1—N2 | 77.15 (10) | C2—C3—H3B | 120.2 |
| O1—P1—O2 | 115.58 (13) | C4—C3—H3B | 120.2 |
| O1—P1—O4 | 110.35 (13) | C3—C4—C5 | 119.8 (3) |
| O2—P1—O4 | 107.98 (14) | C3—C4—H4B | 120.1 |
| O1—P1—O3 | 107.30 (13) | C5—C4—H4B | 120.1 |
| O2—P1—O3 | 108.24 (13) | N1—C5—C4 | 120.9 (3) |
| O4—P1—O3 | 107.05 (14) | N1—C5—C6 | 114.9 (3) |
| O8—P2—O5 | 116.54 (13) | C4—C5—C6 | 124.1 (3) |
| O8—P2—O7 | 112.26 (14) | N2—C6—C7 | 121.0 (3) |
| O5—P2—O7 | 104.93 (13) | N2—C6—C5 | 114.8 (3) |
| O8—P2—O6 | 106.23 (12) | C7—C6—C5 | 124.2 (3) |
| O5—P2—O6 | 108.87 (12) | C8—C7—C6 | 118.7 (4) |
| O7—P2—O6 | 107.72 (14) | C8—C7—H7B | 120.6 |
| P1—O1—Co1 | 129.07 (13) | C6—C7—H7B | 120.6 |
| P1—O3—H3A | 109.5 | C9—C8—C7 | 119.8 (4) |
| P1—O4—H4A | 109.5 | C9—C8—H8A | 120.1 |
| P2—O5—Co1 | 127.55 (12) | C7—C8—H8A | 120.1 |
| P2—O6—H6A | 109.5 | C8—C9—C10 | 118.8 (4) |
| P2—O7—H7A | 109.5 | C8—C9—H9A | 120.6 |
| P2—O8—Co1i | 155.17 (16) | C10—C9—H9A | 120.6 |
| C1—N1—C5 | 118.6 (3) | N2—C10—C9 | 122.6 (4) |
| C1—N1—Co1 | 124.4 (2) | N2—C10—H10A | 118.7 |
| C5—N1—Co1 | 116.9 (2) | C9—C10—H10A | 118.7 |
| C10—N2—C6 | 119.0 (3) | | |
| Symmetry code: (i) −x, −y, −z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O3—H3A···O2ii | 0.82 | 1.71 | 2.522 (4) | 170 |
| O4—H4A···O5 | 0.82 | 1.79 | 2.603 (3) | 170 |
| O6—H6A···O2iii | 0.82 | 1.78 | 2.589 (3) | 169 |
| O7—H7A···O3i | 0.82 | 1.90 | 2.694 (4) | 161 |
| Symmetry codes: (i) −x, −y, −z; (ii) −x+1, −y+1, −z; (iii) −x, −y+1, −z. |
Experimental details
| Crystal data |
| Chemical formula | [Co(H2PO4)4(C10H8N2)2] |
| Mr | 818.17 |
| Crystal system, space group | Triclinic, P1 |
| Temperature (K) | 293 |
| a, b, c (Å) | 8.6797 (17), 8.7749 (18), 10.057 (2) |
| α, β, γ (°) | 96.09 (3), 100.55 (3), 100.89 (3) |
| V (Å3) | 731.6 (3) |
| Z | 1 |
| Radiation type | Mo Kα |
| µ (mm−1) | 1.44 |
| Crystal size (mm) | 0.20 × 0.20 × 0.10 |
| |
| Data collection |
| Diffractometer | Siemens SMART CCD area-detector
diffractometer |
| Absorption correction | Empirical (using intensity measurements)
(SADABS; Sheldrick, 1996) |
| Tmin, Tmax | 0.758, 0.866 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 3435, 2902, 2415 |
| Rint | 0.016 |
| (sin θ/λ)max (Å−1) | 0.622 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.040, 0.091, 1.00 |
| No. of reflections | 2902 |
| No. of parameters | 208 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.41, −0.32 |
Selected geometric parameters (Å, º) top| Co1—O8i | 1.968 (2) | P1—O3 | 1.572 (2) |
| Co1—O1 | 1.993 (2) | P1—O4 | 1.554 (2) |
| Co1—O5 | 2.064 (2) | P2—O5 | 1.505 (2) |
| Co1—N1 | 2.067 (3) | P2—O6 | 1.554 (2) |
| Co1—N2 | 2.117 (3) | P2—O7 | 1.553 (2) |
| P1—O1 | 1.497 (2) | P2—O8 | 1.481 (2) |
| P1—O2 | 1.497 (2) | | |
| | | |
| O8i—Co1—O1 | 108.57 (9) | O5—Co1—N1 | 96.96 (9) |
| O8i—Co1—O5 | 93.73 (9) | O8i—Co1—N2 | 87.81 (10) |
| O1—Co1—O5 | 93.94 (9) | O1—Co1—N2 | 92.14 (10) |
| O8i—Co1—N1 | 134.82 (11) | O5—Co1—N2 | 172.90 (9) |
| O1—Co1—N1 | 114.27 (10) | N1—Co1—N2 | 77.15 (10) |
| Symmetry code: (i) −x, −y, −z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O3—H3A···O2ii | 0.82 | 1.71 | 2.522 (4) | 170 |
| O4—H4A···O5 | 0.82 | 1.79 | 2.603 (3) | 170 |
| O6—H6A···O2iii | 0.82 | 1.78 | 2.589 (3) | 169 |
| O7—H7A···O3i | 0.82 | 1.90 | 2.694 (4) | 161 |
| Symmetry codes: (i) −x, −y, −z; (ii) −x+1, −y+1, −z; (iii) −x, −y+1, −z. |


 journal menu
journal menu metal-organic compounds
metal-organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2004/07/00/na1662/na1662fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2004/07/00/na1662/na1662fig2thm.gif)




Metal phosphonate or ortho-phosphate materials are of increasing interest due to their potential applications in ion exchange, catalysis and sensing (Liang et al., 2003; Wang et al., 2002). An extraordinarily large number of orthophosphates have been characterized in the past two decades (Wilson et al., 1982; Gier & Stucky, 1991). Here, we report the crystal structure of the title compound, (I). \sch
The structure of (I) contains two CoII ions, two 2,2'-bipyridine ligands and four dihydrogen-orthophosphate anions. It is built up of centrosymmetric dinuclear entities. The coordination sphere of the CoII ions is best described as a distorted square pyramid. The basal coordination positions are occupied by two N atoms and two O atoms belonging to two bridging dihydrogen-orthophosphate anions. The axial position is occupied by an O atom from the terminal dihydrogen-orthophosphate anions.
The 2,2'-bipyridine ligands chelate the Co atom and form a five-membered CoN2C2 ring. The Co—N bond lengths of 2.067 (3) and 2.117 (3) Å are in good agreement with the corresponding five-coordination Co—N distances reported previously (Du et al., 2001). The N1—Co1—N2 bite angle is 77.15 (10)°, which is narrower than that in [Co(phen)2(H2O)2](NO3)3·2H2O [84.5 (1)°; Ye et al., 1994].
The two Co atoms are bridged by two orthophosphate anions. The O atoms are bound asymmetrically to Co, with distances of 1.968 (2) and 2.064 (2) Å. The axial Co—O bond length is 1.993 (2) Å. The Co—O bond lengths in this structure are slightly longer than found in a similar structure (Murugavel et al., 2001).
The five-membered chelate ring (P1) is fairly planar, the displacement of atom Co1 from the weighted least-squares plane through N1/C5/C6/N2 being 0.029 (2) Å. The dihedral angles between P1 and the planes N2/C6—C10 (P2) and N1/C1—C5 (P3) are 5.3 (1) and 4.7 (1)°, respectively. Planes P2 and P3 form a dihedral angle of 8.5 (1)°.
There is one intermolecular O—H···O hydrogen bond interaction in the crystal of (I) (Table 2), and these hydrogen-bond interactions stabilize the structure.