Buy article online - an online subscription or single-article purchase is required to access this article.
In the title compound, C
6H
18N
22+·C
4H
4O
42-·C
4H
6O
4, the components lie on centres of symmetry in space group
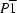
, such that the asymmetric unit contains three half-molecules. Despite the different mode (with respect to other dicarboxylic acids) adopted by the intermolecular self-interaction of succinic acid derivatives, the overall structure of the title compound consists of anionic layers that are typical of the packing structures exhibited by other dicarboxylic acid analogues.
Supporting information
CCDC reference: 237950
Crystals of (I) were obtained by stirring, at room temperature, a 50 mM solution (6 ml) of N,N,N',N'-tetramethyletylenediammine with the same volume of an equimolecular (1:1 ratio) aqueous succinic acid solution. The final stoichiometry of the crystal was 1:2, probably as a result of the acid and base strengths; diammonium salts usually crystallize with semi-carboxylate ions of dicarboxylic acids (Barnes & Weakley, 2000), resulting in the overall ratio 1:2. The mother solution, containing the remaining ammine, was filtered, and samples suitable for X-ray analysis were selected from the dried crystals.
All non-H atoms were located by the SIR97 software for structural solution, whereas H atoms were located in difference Fourier maps. H atoms were then placed in idealized positions (O—H = 0.82 Å, N—H = 0.91 Å and C–H = 0.96 Å) and allowed to ride on their parent atoms, with fixed isotropic displacement parameters (0.05 Å2).
Data collection: XSCANS (Siemens, 1989); cell refinement: XSCANS; data reduction: XPREPW (Bruker, 1997); program(s) used to solve structure: SIR97 (Altomare et al., 1994); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: XPW (Bruker, 1997); software used to prepare material for publication: PARST97 (Nardelli, 1995) and WinGX (Farrugia, 1999).
N,
N,
N',
N'-Tetramethyldiammonium–succinate–succinic acid (1/1/1)
top
Crystal data top
| C6H18N22+·C4H6O42−·C4H4O4 | Z = 1 |
| Mr = 352.38 | F(000) = 190 |
| Triclinic, P1 | Dx = 1.372 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation, λ = 0.71073 Å |
| a = 5.6370 (4) Å | Cell parameters from 46 reflections |
| b = 8.7100 (8) Å | θ = 7.1–17.4° |
| c = 8.8431 (6) Å | µ = 0.11 mm−1 |
| α = 96.219 (7)° | T = 298 K |
| β = 93.414 (6)° | Irregular, colourless |
| γ = 97.604 (7)° | 0.40 × 0.32 × 0.18 mm |
| V = 426.63 (6) Å3 | |
Data collection top
Siemens P4
diffractometer | Rint = 0.015 |
| 2θ/ω scans | θmax = 30°, θmin = 2.3° |
Absorption correction: empirical (using intensity measurements)
(North et al., 1968) | h = −7→1 |
| Tmin = 0.273, Tmax = 0.299 | k = −12→12 |
| 3194 measured reflections | l = −12→12 |
| 2495 independent reflections | 3 standard reflections every 197 reflections |
| 1926 reflections with I > 2σ(I) | intensity decay: 1% |
Refinement top
| Refinement on F2 | 0 restraints |
| Least-squares matrix: full | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.042 | w = 1/[σ2(Fo2) + (0.0553P)2 + 0.073P]
where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.117 | (Δ/σ)max < 0.001 |
| S = 1.06 | Δρmax = 0.32 e Å−3 |
| 2495 reflections | Δρmin = −0.21 e Å−3 |
| 109 parameters | |
Crystal data top
| C6H18N22+·C4H6O42−·C4H4O4 | γ = 97.604 (7)° |
| Mr = 352.38 | V = 426.63 (6) Å3 |
| Triclinic, P1 | Z = 1 |
| a = 5.6370 (4) Å | Mo Kα radiation |
| b = 8.7100 (8) Å | µ = 0.11 mm−1 |
| c = 8.8431 (6) Å | T = 298 K |
| α = 96.219 (7)° | 0.40 × 0.32 × 0.18 mm |
| β = 93.414 (6)° | |
Data collection top
Siemens P4
diffractometer | 1926 reflections with I > 2σ(I) |
Absorption correction: empirical (using intensity measurements)
(North et al., 1968) | Rint = 0.015 |
| Tmin = 0.273, Tmax = 0.299 | 3 standard reflections every 197 reflections |
| 3194 measured reflections | intensity decay: 1% |
| 2495 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.042 | 0 restraints |
| wR(F2) = 0.117 | H-atom parameters constrained |
| S = 1.06 | Δρmax = 0.32 e Å−3 |
| 2495 reflections | Δρmin = −0.21 e Å−3 |
| 109 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| O1 | 0.60797 (19) | 0.31233 (14) | 1.15237 (12) | 0.0532 (3) | |
| H1C | 0.5554 | 0.2808 | 1.0645 | 0.05* | |
| O2 | 0.90969 (19) | 0.47454 (13) | 1.26932 (11) | 0.0473 (3) | |
| C1 | 0.8808 (2) | 0.44766 (15) | 0.99564 (13) | 0.0327 (3) | |
| H1B | 0.8929 | 0.3500 | 0.9345 | 0.05* | |
| H1A | 0.7590 | 0.4970 | 0.9449 | 0.05* | |
| C2 | 0.8032 (2) | 0.41383 (15) | 1.15107 (14) | 0.0327 (3) | |
| O3 | 0.41548 (18) | 0.20430 (14) | 0.89223 (11) | 0.0532 (3) | |
| O4 | 0.10688 (16) | 0.07705 (11) | 0.74727 (10) | 0.0385 (2) | |
| C3 | 0.1199 (2) | 0.05140 (15) | 1.01490 (13) | 0.0336 (3) | |
| H3A | 0.2352 | −0.0079 | 1.0586 | 0.05* | |
| H3B | 0.1038 | 0.1385 | 1.0900 | 0.05* | |
| C4 | 0.2185 (2) | 0.11487 (14) | 0.87434 (13) | 0.0299 (3) | |
| N1 | 0.38054 (17) | 0.19377 (11) | 0.53328 (10) | 0.0261 (2) | |
| H1 | 0.3057 | 0.1659 | 0.6166 | 0.05* | |
| C7 | 0.2093 (2) | 0.26764 (15) | 0.43874 (15) | 0.0346 (3) | |
| H7A | 0.0707 | 0.1928 | 0.4044 | 0.05* | |
| H7B | 0.2862 | 0.3025 | 0.3521 | 0.05* | |
| H7C | 0.1610 | 0.3551 | 0.4989 | 0.05* | |
| C5 | 0.4401 (2) | 0.04896 (13) | 0.44679 (13) | 0.0292 (2) | |
| H5A | 0.5468 | 0.0765 | 0.3689 | 0.05* | |
| H5B | 0.2944 | −0.0115 | 0.3965 | 0.05* | |
| C6 | 0.5959 (2) | 0.30841 (15) | 0.58824 (15) | 0.0358 (3) | |
| H6A | 0.7044 | 0.2598 | 0.6491 | 0.05* | |
| H6B | 0.5479 | 0.3958 | 0.6487 | 0.05* | |
| H6C | 0.6745 | 0.3437 | 0.5024 | 0.05* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| O1 | 0.0452 (6) | 0.0763 (8) | 0.0306 (5) | −0.0228 (5) | 0.0061 (4) | 0.0091 (5) |
| O2 | 0.0485 (6) | 0.0621 (6) | 0.0262 (5) | −0.0070 (5) | −0.0011 (4) | 0.0018 (4) |
| C1 | 0.0294 (6) | 0.0411 (6) | 0.0247 (5) | −0.0059 (5) | 0.0009 (4) | 0.0045 (5) |
| C2 | 0.0305 (6) | 0.0399 (6) | 0.0268 (5) | 0.0003 (5) | 0.0034 (4) | 0.0054 (5) |
| O3 | 0.0420 (5) | 0.0742 (7) | 0.0337 (5) | −0.0307 (5) | 0.0021 (4) | 0.0109 (5) |
| O4 | 0.0367 (5) | 0.0518 (6) | 0.0229 (4) | −0.0112 (4) | 0.0032 (3) | 0.0070 (4) |
| C3 | 0.0303 (6) | 0.0443 (7) | 0.0232 (5) | −0.0100 (5) | 0.0030 (4) | 0.0079 (5) |
| C4 | 0.0284 (6) | 0.0347 (6) | 0.0255 (5) | −0.0036 (5) | 0.0062 (4) | 0.0057 (4) |
| N1 | 0.0286 (5) | 0.0274 (4) | 0.0215 (4) | −0.0003 (4) | 0.0050 (3) | 0.0035 (3) |
| C7 | 0.0356 (6) | 0.0355 (6) | 0.0338 (6) | 0.0073 (5) | 0.0020 (5) | 0.0065 (5) |
| C5 | 0.0342 (6) | 0.0293 (5) | 0.0234 (5) | 0.0040 (4) | 0.0005 (4) | 0.0015 (4) |
| C6 | 0.0361 (6) | 0.0324 (6) | 0.0348 (6) | −0.0077 (5) | 0.0016 (5) | 0.0017 (5) |
Geometric parameters (Å, º) top
| O1—C2 | 1.319 (1) | N1—C6 | 1.487 (1) |
| O1—H1C | 0.82 | N1—C5 | 1.491 (1) |
| O2—C2 | 1.206 (1) | N1—C7 | 1.493 (2) |
| C1—C1i | 1.514 (2) | N1—H1 | 0.91 |
| C1—C2 | 1.514 (2) | C7—H7A | 0.96 |
| C1—H1B | 0.97 | C7—H7B | 0.96 |
| C1—H1A | 0.97 | C7—H7C | 0.96 |
| O3—C4 | 1.260 (1) | C5—C5iii | 1.523 (2) |
| O4—C4 | 1.244 (1) | C5—H5A | 0.97 |
| C3—C3ii | 1.511 (2) | C5—H5B | 0.97 |
| C3—C4 | 1.520 (1) | C6—H6A | 0.96 |
| C3—H3A | 0.97 | C6—H6B | 0.96 |
| C3—H3B | 0.97 | C6—H6C | 0.96 |
| | | |
| C2—O1—H1C | 109.5 | C6—N1—H1 | 107.6 |
| C1i—C1—C2 | 112.76 (12) | C5—N1—H1 | 107.6 |
| C1i—C1—H1B | 109 | C7—N1—H1 | 107.6 |
| C2—C1—H1B | 109 | N1—C7—H7A | 109.5 |
| C1i—C1—H1A | 109 | N1—C7—H7B | 109.5 |
| C2—C1—H1A | 109 | H7A—C7—H7B | 109.5 |
| H1B—C1—H1A | 107.8 | N1—C7—H7C | 109.5 |
| O2—C2—O1 | 120.2 (1) | H7A—C7—H7C | 109.5 |
| O2—C2—C1 | 123.4 (1) | H7B—C7—H7C | 109.5 |
| O1—C2—C1 | 116.3 (1) | N1—C5—C5iii | 110.72 (11) |
| C3ii—C3—C4 | 114.43 (12) | N1—C5—H5A | 109.5 |
| C3ii—C3—H3A | 108.7 | C5iii—C5—H5A | 109.5 |
| C4—C3—H3A | 108.7 | N1—C5—H5B | 109.5 |
| C3ii—C3—H3B | 108.7 | C5iii—C5—H5B | 109.5 |
| C4—C3—H3B | 108.7 | H5A—C5—H5B | 108.1 |
| H3A—C3—H3B | 107.6 | N1—C6—H6A | 109.5 |
| O4—C4—O3 | 122.3 (1) | N1—C6—H6B | 109.5 |
| O4—C4—C3 | 120.1 (1) | H6A—C6—H6B | 109.5 |
| O3—C4—C3 | 117.5 (1) | N1—C6—H6C | 109.5 |
| C6—N1—C5 | 113.13 (9) | H6A—C6—H6C | 109.5 |
| C6—N1—C7 | 110.20 (9) | H6B—C6—H6C | 109.5 |
| C5—N1—C7 | 110.34 (9) | | |
| | | |
| C1i—C1—C2—O2 | 6.0 (2) | C3ii—C3—C4—O3 | 178.5 (1) |
| C1i—C1—C2—O1 | −174.2 (1) | C6—N1—C5—C5iii | 70.7 (1) |
| C3ii—C3—C4—O4 | −2.3 (2) | C7—N1—C5—C5iii | −165.33 (12) |
| Symmetry codes: (i) −x+2, −y+1, −z+2; (ii) −x, −y, −z+2; (iii) −x+1, −y, −z+1. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···O4 | 0.91 | 1.82 | 2.710 (1) | 166 |
| N1—H1···O3 | 0.91 | 2.45 | 3.159 (1) | 134 |
| O1—H1C···O3 | 0.82 | 1.70 | 2.514 (1) | 173 |
| C5—H5B···O4iv | 0.97 | 2.49 | 3.424 (1) | 161 |
| C7—H7C···O2v | 0.96 | 2.48 | 3.392 (2) | 158 |
| Symmetry codes: (iv) −x, −y, −z+1; (v) −x+1, −y+1, −z+2. |
Experimental details
| Crystal data |
| Chemical formula | C6H18N22+·C4H6O42−·C4H4O4 |
| Mr | 352.38 |
| Crystal system, space group | Triclinic, P1 |
| Temperature (K) | 298 |
| a, b, c (Å) | 5.6370 (4), 8.7100 (8), 8.8431 (6) |
| α, β, γ (°) | 96.219 (7), 93.414 (6), 97.604 (7) |
| V (Å3) | 426.63 (6) |
| Z | 1 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.11 |
| Crystal size (mm) | 0.40 × 0.32 × 0.18 |
| |
| Data collection |
| Diffractometer | Siemens P4
diffractometer |
| Absorption correction | Empirical (using intensity measurements)
(North et al., 1968) |
| Tmin, Tmax | 0.273, 0.299 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 3194, 2495, 1926 |
| Rint | 0.015 |
| (sin θ/λ)max (Å−1) | 0.703 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.042, 0.117, 1.06 |
| No. of reflections | 2495 |
| No. of parameters | 109 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.32, −0.21 |
Selected geometric parameters (Å, º) top| O1—C2 | 1.319 (1) | C3—C3ii | 1.511 (2) |
| O2—C2 | 1.206 (1) | C3—C4 | 1.520 (1) |
| C1—C1i | 1.514 (2) | N1—C6 | 1.487 (1) |
| C1—C2 | 1.514 (2) | N1—C5 | 1.491 (1) |
| O3—C4 | 1.260 (1) | N1—C7 | 1.493 (2) |
| O4—C4 | 1.244 (1) | | |
| | | |
| O2—C2—O1 | 120.2 (1) | O4—C4—O3 | 122.3 (1) |
| O2—C2—C1 | 123.4 (1) | O4—C4—C3 | 120.1 (1) |
| O1—C2—C1 | 116.3 (1) | O3—C4—C3 | 117.5 (1) |
| | | |
| C1i—C1—C2—O2 | 6.0 (2) | C3ii—C3—C4—O4 | −2.3 (2) |
| C1i—C1—C2—O1 | −174.2 (1) | C3ii—C3—C4—O3 | 178.5 (1) |
| Symmetry codes: (i) −x+2, −y+1, −z+2; (ii) −x, −y, −z+2. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···O4 | 0.91 | 1.82 | 2.710 (1) | 166 |
| N1—H1···O3 | 0.91 | 2.45 | 3.159 (1) | 134 |
| O1—H1C···O3 | 0.82 | 1.70 | 2.514 (1) | 173 |
| C5—H5B···O4iii | 0.97 | 2.49 | 3.424 (1) | 161 |
| C7—H7C···O2iv | 0.96 | 2.48 | 3.392 (2) | 158 |
| Symmetry codes: (iii) −x, −y, −z+1; (iv) −x+1, −y+1, −z+2. |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2004/04/00/na1646/na1646fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2004/04/00/na1646/na1646fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2004/04/00/na1646/na1646fig3thm.gif)

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry



In the course of a wide-ranging study concerning the solid-state aggregation of anions derived from dicarboxylic acids crystallized with solvents and/or positively charged counter-ions, we have crystallized and solved the structure of an interesting supramolecular solid, (I), formed by succinic acid with an alkyl diammonium succinate. Many studies of the behaviour in the solid state of dicarboxylic acids have been carried out (MacDonald et al. 2001; Thalladi et al., 2000) in order to investigate their trend towards self-aggregation, which stems from the hydrogen interactions (Aakeroy & Seddon, 1993), that these systems are able to develop among themselves. Therefore, dicarboxylic acids provide? suitable substrates for crystal engineering (Desiraiju, 1995, 2003). Succinic acid can be considered the first in the series of dicarboxylic acids whose the two functional groups do not interact sterically with one another unless they are constrained in the symmetric cis conformation (e.g. Sobota & Szafert, 1996). Recently, the structure of this acid (related to its physical behaviour) was analyzed and compared with corresponding superior homologues? (Pedireddi et al., 1998; Thalladi et al. 2000). Moreover, its importance is evident when comsidering the many reported structures with nitrogenated compounds (Kalsbeek, 1991; Kalsbeek & Larsen, 1991; MacDonald et al., 2001; Büyükgüngör & Mustafa, 2002) and amino acids thought to be present in prebiotic conditions (Prasad & Vijayan, 1993; Prasad & Vijayan, 1991). Linear polyamines also form, in aqueous solution, fairly stable complexes with carboxylic ligands, such as dicarboxylates (malonates, succinates, etc.; De Robertis et al., 2001, and references therein), and these interactions can be considered as useful models for the binding of polyanions by polyammonium cations in natural systems.
The asymmetric unit of (I) contains one-half of a succinic acid molecule, one-half of a succinate anion and one-half of an N,N,N',N'-tetramethyletylenediammonium cation (Fig. 1). As expected, the succinic fragments are planar [the maximum deviations are for atoms C2, by 0.001 (1) Å, and C4, by 0.004 (1) Å], but they are also coplanar [the angle between their mean planes is 8.82 (5)°]. This disposition is driven by the stable planar conformation of the succinic derivatives, combined with the development of directional hydrogen-bonding interactions employing the lone pairs of carboxylate atoms O3 and O4 (anti towards the OH donors and syn towards NH). Succinic acid and succinate molecules, bound via an intermolecular hydrogen bond (H1C···O3 = 1.70 Å), alternate along one-dimensional ribbons parallel to the ab crystallographic plane and along the [110] direction (Fig. 2).
In the carboxy moieties, O-atom lone pairs and/or H atoms can be placed on the same side as the other O atom (syn) or on the opposite side (anti), so it is straightforward to infer that hydrogen-bonding interactions can be developed towards these two possible directions, either for O-donors or for O-acceptors. Usually the syn positions are less sterically hindered and thus are often exploited by strong molecular interactions and/or synthons (Fleishman et al., 2003). A search of the Cambridge Structural Database (Allen, 2002) for any ammonium succinate showns that this dicarboxylic acid often self-assesembles along one-dimensional ribbons or strands (planar or not), obtained through different hydrogen-bonding pathways, viz. anti–anti (three examples), syn–syn (14 examples) and syn–anti (four examples). The structure of (I) confirms that succinic derivates are able to self-aggregate via anti–anti (Fig. 2) interactions, unlike all of the previously reported dicarboxylic acids (MacDonald & al., 2001), which adopt the more common syn–syn hydrogen bond for self-aggregation. We suggest that, in this way, succinate networks free their stronger O-donor syn sites, which are then able to bind tightly to specific guest counter-ions. To our knowledge, there is only one pecedent, in which the acid presents two highly symmetric ionization states that lead to this particular ribbon network (YOWDET; Prasad & Vijayan, 1990). Therefore, we are currently studying the possible influence of different counter-ions on the succinic self-assembly mode. In (I), counteri-ons bear positively charged mono-hydrogenated N atoms, which lie close to the mean plane of the succinate molecule [the deviation from the plane is 0.272 (1) Å], so we might infer that this interaction between the two oppositely charged ions is directional. This strong hydrogen bond (H1···O4 = 1.82 Å), together with some weaker bonds (Table 2), holds the succinic ribbons side-by-side, such that they appear as supermolecular layers. These layers are, in turn, connected along the third dimension by ethylenediammonic chains (see Fig. 3).