In the crystal structure of the title compound, C
14H
12N
2O
2, the molecule lies about a twofold axis; two carbonyl groups and the H atoms of the N—N bond are in a
trans orientation with respect to each other. In the crystal, each molecule is linked to the other and
vice versa by intermolecular N—H

O hydrogen bonds between the amide hydrogen and the O atoms of neighbouring molecules to form two ten-membered rings, each of which has the graph-set motif
C4
R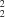
(10). This extends as a polymeric chain along the
c axis.
Supporting information
CCDC reference: 150350
The compound was prepared by adding benzoylchloride into the mixture of hydrazine, potassium hydroxide and carbon disulfide in 20% aqueous ethanol at 273 K. The precipitate was washed with water and dried. The crystal was obtained by recrystallization from ethanol.
Data collection was as that followed by Shanmuga Sundara Raj et al. (1999).
Data collection: SMART (Siemens, 1996); cell refinement: SAINT; data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 1997); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and PARST (Nardelli, 1995).
1,2,dibenzoylhydrazine
top
Crystal data top
| C14H12N2O2 | F(000) = 504 |
| Mr = 240.26 | Dx = 1.339 Mg m−3 |
| Monoclinic, C2/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 14.5439 (2) Å | Cell parameters from 2038 reflections |
| b = 9.7314 (3) Å | θ = 3.0–28.3° |
| c = 9.0181 (3) Å | µ = 0.09 mm−1 |
| β = 110.938 (1)° | T = 293 K |
| V = 1192.07 (6) Å3 | Block, light yellow |
| Z = 4 | 0.32 × 0.30 × 0.28 mm |
Data collection top
Siemens SMART CCD area detector
diffractometer | 965 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.054 |
| Graphite monochromator | θmax = 28.3°, θmin = 3.0° |
| Detector resolution: 8.33 pixels mm-1 | h = −19→14 |
| ω scans | k = −12→12 |
| 4148 measured reflections | l = −10→12 |
| 1471 independent reflections | |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.053 | H-atom parameters constrained |
| wR(F2) = 0.164 | w = 1/[σ2(Fo2) + (0.0975P)2]
where P = (Fo2 + 2Fc2)/3 |
| S = 0.96 | (Δ/σ)max < 0.001 |
| 1471 reflections | Δρmax = 0.30 e Å−3 |
| 83 parameters | Δρmin = −0.28 e Å−3 |
| 0 restraints | Extinction correction: SHELXTL (Sheldrick, 1997), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.021 (5) |
Crystal data top
| C14H12N2O2 | V = 1192.07 (6) Å3 |
| Mr = 240.26 | Z = 4 |
| Monoclinic, C2/c | Mo Kα radiation |
| a = 14.5439 (2) Å | µ = 0.09 mm−1 |
| b = 9.7314 (3) Å | T = 293 K |
| c = 9.0181 (3) Å | 0.32 × 0.30 × 0.28 mm |
| β = 110.938 (1)° | |
Data collection top
Siemens SMART CCD area detector
diffractometer | 965 reflections with I > 2σ(I) |
| 4148 measured reflections | Rint = 0.054 |
| 1471 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.053 | 0 restraints |
| wR(F2) = 0.164 | H-atom parameters constrained |
| S = 0.96 | Δρmax = 0.30 e Å−3 |
| 1471 reflections | Δρmin = −0.28 e Å−3 |
| 83 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| O1 | 0.07908 (9) | 0.87939 (14) | 0.10828 (13) | 0.0480 (4) | |
| N1 | 0.05005 (10) | 1.00954 (16) | 0.29345 (16) | 0.0449 (5) | |
| H1A | 0.0736 | 1.0562 | 0.3798 | 0.054* | |
| C1 | 0.21547 (11) | 0.92445 (18) | 0.34607 (17) | 0.0364 (4) | |
| C2 | 0.26157 (14) | 1.0190 (2) | 0.4632 (2) | 0.0475 (5) | |
| H2A | 0.2267 | 1.0939 | 0.4796 | 0.057* | |
| C3 | 0.36046 (15) | 1.0019 (2) | 0.5567 (2) | 0.0599 (6) | |
| H3A | 0.3917 | 1.0659 | 0.6351 | 0.072* | |
| C4 | 0.41221 (15) | 0.8902 (2) | 0.5335 (2) | 0.0581 (6) | |
| H4A | 0.4784 | 0.8797 | 0.5954 | 0.070* | |
| C5 | 0.36629 (15) | 0.7951 (2) | 0.4196 (2) | 0.0535 (6) | |
| H5A | 0.4009 | 0.7188 | 0.4059 | 0.064* | |
| C6 | 0.26837 (14) | 0.8120 (2) | 0.32478 (19) | 0.0464 (5) | |
| H6A | 0.2378 | 0.7477 | 0.2463 | 0.056* | |
| C7 | 0.11007 (12) | 0.93454 (18) | 0.23956 (17) | 0.0364 (4) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| O1 | 0.0420 (7) | 0.0675 (9) | 0.0314 (6) | −0.0041 (6) | 0.0094 (5) | −0.0060 (5) |
| N1 | 0.0281 (8) | 0.0723 (11) | 0.0315 (7) | −0.0012 (7) | 0.0073 (6) | −0.0065 (6) |
| C1 | 0.0310 (9) | 0.0493 (10) | 0.0304 (8) | −0.0025 (7) | 0.0129 (7) | 0.0047 (7) |
| C2 | 0.0346 (10) | 0.0607 (12) | 0.0464 (10) | −0.0023 (8) | 0.0136 (8) | −0.0070 (8) |
| C3 | 0.0370 (11) | 0.0832 (15) | 0.0526 (11) | −0.0073 (10) | 0.0075 (9) | −0.0137 (10) |
| C4 | 0.0320 (10) | 0.0893 (16) | 0.0491 (11) | 0.0059 (10) | 0.0097 (8) | 0.0064 (10) |
| C5 | 0.0472 (11) | 0.0663 (13) | 0.0499 (11) | 0.0157 (9) | 0.0208 (9) | 0.0096 (9) |
| C6 | 0.0455 (11) | 0.0544 (11) | 0.0402 (9) | 0.0023 (8) | 0.0164 (8) | 0.0003 (8) |
| C7 | 0.0325 (9) | 0.0482 (9) | 0.0287 (8) | −0.0051 (7) | 0.0111 (6) | 0.0041 (6) |
Geometric parameters (Å, º) top
| O1—C7 | 1.229 (2) | C2—H2A | 0.9300 |
| N1—C7 | 1.354 (2) | C3—C4 | 1.380 (3) |
| N1—N1i | 1.385 (3) | C3—H3A | 0.9300 |
| N1—H1A | 0.8600 | C4—C5 | 1.367 (3) |
| C1—C2 | 1.380 (2) | C4—H4A | 0.9300 |
| C1—C6 | 1.390 (3) | C5—C6 | 1.384 (3) |
| C1—C7 | 1.493 (2) | C5—H5A | 0.9300 |
| C2—C3 | 1.393 (3) | C6—H6A | 0.9300 |
| | | |
| C7—N1—N1i | 118.6 (1) | C5—C4—C3 | 120.06 (18) |
| C7—N1—H1A | 120.7 | C5—C4—H4A | 120.0 |
| N1i—N1—H1A | 120.7 | C3—C4—H4A | 120.0 |
| C2—C1—C6 | 119.26 (16) | C4—C5—C6 | 120.16 (19) |
| C2—C1—C7 | 123.69 (16) | C4—C5—H5A | 119.9 |
| C6—C1—C7 | 117.04 (16) | C6—C5—H5A | 119.9 |
| C1—C2—C3 | 119.86 (18) | C5—C6—C1 | 120.41 (17) |
| C1—C2—H2A | 120.1 | C5—C6—H6A | 119.8 |
| C3—C2—H2A | 120.1 | C1—C6—H6A | 119.8 |
| C4—C3—C2 | 120.22 (19) | O1—C7—N1 | 121.3 (2) |
| C4—C3—H3A | 119.9 | O1—C7—C1 | 122.0 (2) |
| C2—C3—H3A | 119.9 | N1—C7—C1 | 116.7 (1) |
| | | |
| C6—C1—C2—C3 | 0.9 (3) | C7—C1—C6—C5 | −179.59 (15) |
| C7—C1—C2—C3 | −179.67 (16) | N1i—N1—C7—O1 | 7.9 (2) |
| C1—C2—C3—C4 | −0.5 (3) | N1i—N1—C7—C1 | −173.15 (13) |
| C2—C3—C4—C5 | −0.7 (3) | C2—C1—C7—O1 | 156.00 (17) |
| C3—C4—C5—C6 | 1.5 (3) | C6—C1—C7—O1 | −24.6 (2) |
| C4—C5—C6—C1 | −1.1 (3) | C2—C1—C7—N1 | −23.0 (2) |
| C2—C1—C6—C5 | −0.2 (3) | C6—C1—C7—N1 | 156.41 (15) |
| Symmetry code: (i) −x, y, −z+1/2. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···O1ii | 0.86 | 2.13 | 2.924 (2) | 154 |
| Symmetry code: (ii) x, −y+2, z+1/2. |
Experimental details
| Crystal data |
| Chemical formula | C14H12N2O2 |
| Mr | 240.26 |
| Crystal system, space group | Monoclinic, C2/c |
| Temperature (K) | 293 |
| a, b, c (Å) | 14.5439 (2), 9.7314 (3), 9.0181 (3) |
| β (°) | 110.938 (1) |
| V (Å3) | 1192.07 (6) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.09 |
| Crystal size (mm) | 0.32 × 0.30 × 0.28 |
| |
| Data collection |
| Diffractometer | Siemens SMART CCD area detector
diffractometer |
| Absorption correction | – |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 4148, 1471, 965 |
| Rint | 0.054 |
| (sin θ/λ)max (Å−1) | 0.667 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.053, 0.164, 0.96 |
| No. of reflections | 1471 |
| No. of parameters | 83 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.30, −0.28 |
Selected geometric parameters (Å, º) top| O1—C7 | 1.229 (2) | N1—N1i | 1.385 (3) |
| N1—C7 | 1.354 (2) | | |
| | | |
| C7—N1—N1i | 118.6 (1) | O1—C7—C1 | 122.0 (2) |
| O1—C7—N1 | 121.3 (2) | N1—C7—C1 | 116.7 (1) |
| Symmetry code: (i) −x, y, −z+1/2. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···O1ii | 0.86 | 2.13 | 2.924 (2) | 154 |
| Symmetry code: (ii) x, −y+2, z+1/2. |


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2000/08/00/na1474/na1474fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2000/08/00/na1474/na1474fig2thm.gif)




Both S-methyldithiocarbazate and S-benzylcarbazate can be prepared by the addition of chloromethane or benzylchloride into a mixture of hydrazine and carbon disulfide, respectively (Lanfredi et al., 1977; Mattes & Weber, 1980; Ali & Tarafder, 1977). However, under the same conditions 1,2-dibenzoyl hydrazine, (I), was obtained instead of S-benzoyldithiocarbazate when benzoylchloride was added to the reaction mixture. \sch
The dihedral angle of 24.0 (1)° between the phenyl ring and the N1—C7=O1 group shows that the molecule is not planar. The asymmetric unit contains half the molecule and the other half is related by a crystallographic twofold axis passing through N1—N1i bond [(i) = −x, y, 1/2 − z]. The symmetry-related group adopts anti-clinal orientation with respect to the NN bond; the torsion angle, C7—N1—N1i—C7i, being 104.3 (2)°. The two carbonyl groups and hydrogen atoms of the NN bond are in a trans orientation to each other.
The bond lengths and bond angles are comparable with those of N,N'-bis (picolinoyl)hydrazine (Shao et al., 1999) except for C═O which is slightly longer and N—C—C angle [114.7 (1)°] which is 2° larger, may be due to the more twist along the N—Ni bond [torsion angle C—N—Ni—Ci in Shao et al. (1999) is −70.9 (2)°].
In the crystal, each molecule is linked to the other and vice-versa by intermolecular N—H···O hydrogen bondings between the amide hydrogen and the oxygen atom of the neighbouring molecules to form two ten-membered rings each of which has the graph-set motif of C4R22(10) (Bernstein et al., 1995). This extends as a polymeric chain along the c axis.