Buy article online - an online subscription or single-article purchase is required to access this article.
The title compound, hexaammonium tetra-μ
3-selenido-tetrakis(tricyanomolybdenum) hexahydrate, is isostructural with the Mo/S, W/S and W/Se analogues. The structure contains disordered cyclic hydrogen-bonded [{(NH
4)(H
2O)}
3]
3+ cations and [Mo
4Se
4(CN)
12]
6− cluster anions with
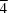
3
m symmetry. The cation assembly consists of alternating ammonium and water molecules linked by N—H

O hydrogen bonds. The anion has a typical cubane cluster structure. The cations and anions are linked together by hydrogen bonds involving the terminal N atoms of the CN groups.
Supporting information
A mixture of Mo3Se7Br4 (1.00 g, 0.862 mmol) and KCN (1.00 g, 15.3 mmol)
was heated in a sealed Pyrex tube at 703 K for 48 h. The product was added to
water (20 ml) and the mixture was refluxed for 1 h and filtered. The potassium
salt isolated by addition of methanol was dissolved in CH3COONH4
(1M, 10 ml) and the mixture was allowed to stand at 293 K for 5–7 d.
Dark red-brown octahedral crystals were isolated by filtration, washed with
methanol and dried in air (yield 0.10 g, 12%). Analysis calculated for
C12H36Mo4N18O6Se4: C 11.74, H 2.95, N 20.53%; found: C 11.54, H
3.00, N 20.20%; IR (KBr): 2132 (CN) cm-1; UV-visible absorption spectrum of
an aqueous solution, λ (ε in M-1cm-1): 340 (9680), 460 (1950) sh,
530 s h, 690 (310) nm.
The largest difference peak is near the anion, 0.6 Å from Se1. The deepest
hole is near the disordered cation, 1.36 Å from H3.
Data collection: SMART (Bruker, 1998); cell refinement: SAINT (Bruker, 1998); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 1998); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and local programs.
hexaammonium tetra-µ
3-selenido-tetrakis[tricyanomolybdenum] hexahydrate
top
Crystal data top
| (NH4)6[Mo4Se4(CN)12]·6H2O | Dx = 2.257 Mg m−3 |
| Mr = 1228.19 | Mo Kα radiation, λ = 0.71073 Å |
| Cubic, Pn3m | Cell parameters from 4887 reflections |
| Hall symbol: -P 4bc 2bc 3 | θ = 2.9–28.6° |
| a = 12.180 (1) Å | µ = 5.44 mm−1 |
| V = 1806.9 (3) Å3 | T = 160 K |
| Z = 2 | Octahedron, black |
| F(000) = 1172 | 0.20 × 0.18 × 0.15 mm |
Data collection top
Siemens SMART CCD
diffractometer | 468 independent reflections |
| Radiation source: fine-focus sealed tube | 384 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.039 |
| Detector resolution: 8.125 pixels mm-1 | θmax = 28.7°, θmin = 2.4° |
| ω rotation with narrow frames scans | h = −7→16 |
Absorption correction: multi-scan
(SADABS; Sheldrick, 1997) | k = −16→15 |
| Tmin = 0.784, Tmax = 0.928 | l = −15→15 |
| 10436 measured reflections | |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.068 | Only H-atom coordinates refined |
| wR(F2) = 0.184 | w = 1/[σ2(Fo2) + (0.0111P)2 + 84.4748P]
where P = (Fo2 + 2Fc2)/3 |
| S = 1.48 | (Δ/σ)max = 0.007 |
| 468 reflections | Δρmax = 0.97 e Å−3 |
| 33 parameters | Δρmin = −1.13 e Å−3 |
| 4 restraints | Extinction correction: SHELXTL (Sheldrick, 1998), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.00004 (14) |
Crystal data top
| (NH4)6[Mo4Se4(CN)12]·6H2O | Z = 2 |
| Mr = 1228.19 | Mo Kα radiation |
| Cubic, Pn3m | µ = 5.44 mm−1 |
| a = 12.180 (1) Å | T = 160 K |
| V = 1806.9 (3) Å3 | 0.20 × 0.18 × 0.15 mm |
Data collection top
Siemens SMART CCD
diffractometer | 468 independent reflections |
Absorption correction: multi-scan
(SADABS; Sheldrick, 1997) | 384 reflections with I > 2σ(I) |
| Tmin = 0.784, Tmax = 0.928 | Rint = 0.039 |
| 10436 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.068 | 4 restraints |
| wR(F2) = 0.184 | Only H-atom coordinates refined |
| S = 1.48 | w = 1/[σ2(Fo2) + (0.0111P)2 + 84.4748P]
where P = (Fo2 + 2Fc2)/3 |
| 468 reflections | Δρmax = 0.97 e Å−3 |
| 33 parameters | Δρmin = −1.13 e Å−3 |
Special details top
Experimental. none |
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R-factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | Occ. (<1) |
| Mo1 | 0.33379 (12) | 0.33379 (12) | 0.16621 (12) | 0.0184 (7) | |
| Se1 | 0.13358 (15) | 0.36642 (15) | 0.13358 (15) | 0.0191 (7) | |
| C1 | 0.3450 (11) | 0.5115 (16) | 0.1550 (11) | 0.031 (4) | |
| N1 | 0.3495 (12) | 0.6041 (14) | 0.1505 (12) | 0.046 (5) | |
| O1 | 0.1056 (9) | 0.6695 (14) | 0.1056 (9) | 0.033 (3) | 0.50 |
| N2 | 0.1056 (9) | 0.6695 (14) | 0.1056 (9) | 0.033 (3) | 0.50 |
| H1 | 0.117 (11) | 0.749 (4) | 0.117 (11) | 0.030* | |
| H2 | 0.029 (9) | 0.64 (2) | 0.116 (9) | 0.030* | 0.50 |
| H3 | 0.159 (4) | 0.641 (15) | 0.159 (4) | 0.030* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Mo1 | 0.0184 (7) | 0.0184 (7) | 0.0184 (7) | −0.0006 (6) | 0.0006 (6) | 0.0006 (6) |
| Se1 | 0.0191 (7) | 0.0191 (7) | 0.0191 (7) | 0.0005 (7) | −0.0005 (7) | 0.0005 (7) |
| C1 | 0.031 (6) | 0.032 (10) | 0.031 (6) | −0.013 (5) | 0.005 (8) | 0.013 (5) |
| N1 | 0.057 (8) | 0.024 (9) | 0.057 (8) | 0.006 (6) | 0.005 (11) | −0.006 (6) |
| O1 | 0.035 (5) | 0.028 (8) | 0.035 (5) | −0.001 (5) | −0.002 (7) | −0.001 (5) |
| N2 | 0.035 (5) | 0.028 (8) | 0.035 (5) | −0.001 (5) | −0.002 (7) | −0.001 (5) |
Geometric parameters (Å, º) top
| Mo1—Mo1i | 2.886 (4) | Mo1—C1v | 2.17 (2) |
| Mo1—Mo1ii | 2.887 (4) | Se1—Mo1i | 2.502 (3) |
| Mo1—Mo1iii | 2.887 (4) | Se1—Mo1ii | 2.502 (3) |
| Mo1—Se1 | 2.502 (3) | C1—N1 | 1.13 (2) |
| Mo1—Se1i | 2.502 (3) | O1—H1 | 0.99 (3) |
| Mo1—Se1ii | 2.503 (3) | O1—H2 | 0.99 (3) |
| Mo1—C1 | 2.17 (2) | O1—H3 | 0.99 (3) |
| Mo1—C1iv | 2.17 (2) | | |
| | | |
| Mo1i—Mo1—Mo1ii | 60.0 | C1—Mo1—Mo1iii | 138.5 (3) |
| Mo1i—Mo1—Mo1iii | 60.0 | C1v—Mo1—Mo1iii | 95.1 (5) |
| Mo1ii—Mo1—Mo1iii | 60.0 | C1iv—Mo1—Se1i | 83.9 (3) |
| Se1i—Mo1—Mo1i | 54.78 (6) | C1—Mo1—Se1i | 161.9 (5) |
| Se1—Mo1—Mo1i | 54.78 (6) | C1v—Mo1—Se1i | 83.9 (3) |
| Se1ii—Mo1—Mo1i | 102.98 (7) | C1iv—Mo1—Se1 | 83.9 (3) |
| Se1i—Mo1—Mo1ii | 102.98 (7) | C1—Mo1—Se1 | 83.9 (3) |
| Se1—Mo1—Mo1ii | 54.78 (6) | C1v—Mo1—Se1 | 161.9 (5) |
| Se1ii—Mo1—Mo1ii | 54.78 (6) | C1iv—Mo1—Se1ii | 161.9 (5) |
| Se1i—Mo1—Mo1iii | 54.78 (6) | C1—Mo1—Se1ii | 83.9 (3) |
| Se1—Mo1—Mo1iii | 102.98 (7) | C1v—Mo1—Se1ii | 83.9 (3) |
| Se1ii—Mo1—Mo1iii | 54.78 (6) | C1iv—Mo1—C1 | 82.6 (8) |
| Se1i—Mo1—Se1 | 106.52 (8) | C1iv—Mo1—C1v | 82.6 (8) |
| Se1i—Mo1—Se1ii | 106.52 (8) | C1—Mo1—C1v | 82.6 (8) |
| Se1—Mo1—Se1ii | 106.52 (8) | Mo1i—Se1—Mo1 | 70.4 (1) |
| C1iv—Mo1—Mo1i | 95.1 (5) | Mo1i—Se1—Mo1ii | 70.4 (1) |
| C1—Mo1—Mo1i | 138.5 (3) | Mo1—Se1—Mo1ii | 70.4 (1) |
| C1v—Mo1—Mo1i | 138.5 (3) | N1—C1—Mo1 | 179 (2) |
| C1iv—Mo1—Mo1ii | 138.5 (3) | H1—O1—H2 | 116 (10) |
| C1—Mo1—Mo1ii | 95.1 (5) | H1—O1—H3 | 99 (10) |
| C1v—Mo1—Mo1ii | 138.5 (3) | H2—O1—H3 | 115 (10) |
| C1iv—Mo1—Mo1iii | 138.5 (3) | | |
| Symmetry codes: (i) −x+1/2, −y+1/2, z; (ii) −x+1/2, y, −z+1/2; (iii) x, −y+1/2, −z+1/2; (iv) −z+1/2, x, −y+1/2; (v) y, −z+1/2, −x+1/2. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···N1vi | 0.99 (3) | 1.88 (4) | 2.86 (3) | 174 (17) |
| O1—H2···O1vii | 0.99 (3) | 1.82 (3) | 2.80 (2) | 166 (5) |
| Symmetry codes: (vi) −x+1/2, −y+3/2, z; (vii) −z, x+1/2, y−1/2. |
Experimental details
| Crystal data |
| Chemical formula | (NH4)6[Mo4Se4(CN)12]·6H2O |
| Mr | 1228.19 |
| Crystal system, space group | Cubic, Pn3m |
| Temperature (K) | 160 |
| a (Å) | 12.180 (1) |
| V (Å3) | 1806.9 (3) |
| Z | 2 |
| Radiation type | Mo Kα |
| µ (mm−1) | 5.44 |
| Crystal size (mm) | 0.20 × 0.18 × 0.15 |
| |
| Data collection |
| Diffractometer | Siemens SMART CCD
diffractometer |
| Absorption correction | Multi-scan
(SADABS; Sheldrick, 1997) |
| Tmin, Tmax | 0.784, 0.928 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 10436, 468, 384 |
| Rint | 0.039 |
| (sin θ/λ)max (Å−1) | 0.675 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.068, 0.184, 1.48 |
| No. of reflections | 468 |
| No. of parameters | 33 |
| No. of restraints | 4 |
| H-atom treatment | Only H-atom coordinates refined |
| w = 1/[σ2(Fo2) + (0.0111P)2 + 84.4748P]
where P = (Fo2 + 2Fc2)/3 |
| Δρmax, Δρmin (e Å−3) | 0.97, −1.13 |
Selected geometric parameters (Å, º) top| Mo1—Mo1i | 2.886 (4) | Mo1—C1 | 2.17 (2) |
| Mo1—Se1 | 2.502 (3) | C1—N1 | 1.13 (2) |
| | | |
| Se1—Mo1—Mo1i | 54.78 (6) | C1—Mo1—Mo1iii | 95.1 (5) |
| Se1—Mo1—Mo1ii | 102.98 (7) | C1—Mo1—C1iv | 82.6 (8) |
| Se1—Mo1—Se1iii | 106.52 (8) | Mo1—Se1—Mo1iii | 70.4 (1) |
| C1—Mo1—Mo1i | 138.5 (3) | N1—C1—Mo1 | 179 (2) |
| Symmetry codes: (i) −x+1/2, −y+1/2, z; (ii) x, −y+1/2, −z+1/2; (iii) −x+1/2, y, −z+1/2; (iv) y, −z+1/2, −x+1/2. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···N1v | 0.99 (3) | 1.88 (4) | 2.86 (3) | 174 (17) |
| O1—H2···O1vi | 0.99 (3) | 1.82 (3) | 2.80 (2) | 166 (5) |
| Symmetry codes: (v) −x+1/2, −y+3/2, z; (vi) −z, x+1/2, y−1/2. |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu inorganic compounds
inorganic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2000/03/00/gs1060/gs1060fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2000/03/00/gs1060/gs1060fig2thm.gif)

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry



Cuboidal clusters with bridging chalcogenide ligands are known for a wide variety of transition metals (Holm, 1992; Coucouvanis, 1991; Saito, 1995, 1997; Shibahara, 1991, 1993; Müller, 1986). Previously, we have prepared and structurally investigated some salts of alkali metal cations and cyanide cluster anions of the general formula [M4Q4(CN)12]n-, where M = Mo, Q = Te, or M = W, Q = S, Se or Te (Fedin, Kalinina et al., 1999). All these compounds belong to triclinic, monoclinic or orthorhombic crystal systems and the anions are slightly distorted from ideal Td point symmetry.
Recently, we have found that Mo/W/S/Se anions can give high symmetry cubic crystal structures with six ammonium cations and six water molecules. Three of them, for M = Mo/Q = S and M = W/Q = S or Se, were structurally investigated at room temperature (Fedin, Samsonenko et al., 2000). They appeared to be isostructural, the anions having the ideal Td point symmetry for metal and chalcogen atoms. Unfortunately, our attemts to find the H atom positions was unsuccessful, due to the disorder of the cations.
In the present article, we report the synthesis and low temperature crystal structure investigation of (NH4)6[Mo4Se4(CN)12]·6H2O, (I), which is isostructural with the Mo/S, W/S and W/Se analogues.
The structure of (I) contains an unusual [{(NH4)(H2O)}3]3+ cation (Fig 1a). This is a ring of alternating ammonium and water molecules linked by NH···O hydrogen bonds. The ring is not planar; it has a chair conformation. The cation assembly is disordered relative to the pseudo-rotation around the centre of the ring in such a way that each position is statistically occupied by one half of NH4+ and one half of H2O. Therefore, the central atom, O1, has a mixed atomic scattering factor, 0.5 N + 0.5O. Due to the low temperature of the experiment we have found the positions of the H atoms. Two of them, H1 and H3, are fully occupied. They correspond to the water molecule or two of the four H atoms of the NH4+ cations. The position of H2 is half-occupied and corresponds to the other two H atoms of the ammonium cations. The N···O hydrogen-bond distance is 2.80 (2) Å, with an angle of 166 (5)° at the H atom.
The anion (Fig. 1 b) has a typical cubane cluster structure and possesses the highest possible point symmetry, Td. Mo—Mo and Mo—Se distances are close to those found for [W4Se4(CN)12]6- (Fedin, Kalinina et al., 1999; Fedin, Samsonenko et al., 1999).
The cations and anions are linked together by hydrogen bonds involving the terminal N atoms of the CN groups (Fig. 2). For each disordered water/ammonium site, there is a hydrogen bond with O/N···N = 2.86 (3) Å and an angle of 174 (17)° at the H1 atom.