In 5-benzyl-1,7-dimethyl-4,5,6,7-tetrahydro-1
H-pyrazolo[3,4-
d]pyrimidine-4,6-dione, C
14H
14N
4O
2, which crystallizes in space group
P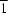
, weak intermolecular C—H

O hydrogen bonds generate dimers. The isomeric compound 1-benzyl-5,7-dimethyl-4,5,6,7-tetrahydro-1
H-pyrazolo[3,4-
d]pyrimidine-4,6-dione, C
14H
14N
4O
2, crystallizes in space group
P2
1/
n, and shows no such dimerization. Instead, it exhibits C—H
π interactions with the phenyl ring. In both structures, the molecules are linked by aromatic π–π-stacking interactions.
Supporting information
CCDC references: 188609; 188610
Compounds (I) and (II) were synthesized as described in the literature (Avasthii
et al.,1998). Diffraction quality crystals of both compounds were
obtained by slow evaporation of ethyl acetate/hexane solutions at room
temperature.
For both compounds, data collection: XSCANS (Siemens, 1996); cell refinement: XSCANS; data reduction: XSCANS; program(s) used to solve structure: SHELXTL-NT (Bruker, 1997); program(s) used to refine structure: SHELXTL-NT; molecular graphics: SHELXTL-NT; software used to prepare material for publication: SHELXTL-NT.
(I) 5-benzyl-1,7-dimethyl-4,6-dioxo-4,5,6,7-tetrahydropyrazolo[3,4-
d]pyrimidine
top
Crystal data top
| C14H14N4O2 | F(000) = 284 |
| Mr = 270.29 | Dx = 1.380 Mg m−3 |
| Triclinic, P1 | Melting point: 165 K |
| a = 7.476 (1) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 8.923 (1) Å | Cell parameters from 59 reflections |
| c = 10.155 (1) Å | θ = 5.0–14.9° |
| α = 76.68 (1)° | µ = 0.10 mm−1 |
| β = 89.08 (1)° | T = 293 K |
| γ = 80.66 (1)° | Block, colourless |
| V = 650.3 (1) Å3 | 0.45 × 0.30 × 0.20 mm |
| Z = 2 | |
Data collection top
Bruker P4
diffractometer | Rint = 0.015 |
| Radiation source: fine-focus sealed tube | θmax = 26.0°, θmin = 2.1° |
| Graphite monochromator | h = −1→9 |
| θ–2θ scans | k = −10→10 |
| 3155 measured reflections | l = −12→12 |
| 2539 independent reflections | 3 standard reflections every 97 reflections |
| 2128 reflections with I > 2σ(I) | intensity decay: none |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.046 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.134 | H-atom parameters constrained |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.0745P)2 + 0.1361P]
where P = (Fo2 + 2Fc2)/3 |
| 2539 reflections | (Δ/σ)max < 0.001 |
| 183 parameters | Δρmax = 0.16 e Å−3 |
| 0 restraints | Δρmin = −0.30 e Å−3 |
Crystal data top
| C14H14N4O2 | γ = 80.66 (1)° |
| Mr = 270.29 | V = 650.3 (1) Å3 |
| Triclinic, P1 | Z = 2 |
| a = 7.476 (1) Å | Mo Kα radiation |
| b = 8.923 (1) Å | µ = 0.10 mm−1 |
| c = 10.155 (1) Å | T = 293 K |
| α = 76.68 (1)° | 0.45 × 0.30 × 0.20 mm |
| β = 89.08 (1)° | |
Data collection top
Bruker P4
diffractometer | Rint = 0.015 |
| 3155 measured reflections | 3 standard reflections every 97 reflections |
| 2539 independent reflections | intensity decay: none |
| 2128 reflections with I > 2σ(I) | |
Refinement top
| R[F2 > 2σ(F2)] = 0.046 | 0 restraints |
| wR(F2) = 0.134 | H-atom parameters constrained |
| S = 1.05 | Δρmax = 0.16 e Å−3 |
| 2539 reflections | Δρmin = −0.30 e Å−3 |
| 183 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| N1 | 0.02330 (18) | 0.88518 (15) | 0.27577 (13) | 0.0451 (3) | |
| N2 | 0.14767 (19) | 0.98266 (16) | 0.22909 (15) | 0.0500 (4) | |
| C3 | 0.2439 (2) | 0.98326 (18) | 0.33610 (17) | 0.0458 (4) | |
| H3 | 0.3378 | 1.0404 | 0.3344 | 0.055* | |
| C3A | 0.18683 (19) | 0.88677 (17) | 0.45331 (16) | 0.0394 (3) | |
| C4 | 0.2426 (2) | 0.84822 (18) | 0.59221 (17) | 0.0420 (4) | |
| N5 | 0.14501 (17) | 0.74149 (15) | 0.67411 (13) | 0.0412 (3) | |
| C6 | 0.0018 (2) | 0.68083 (18) | 0.63141 (16) | 0.0424 (4) | |
| N7 | −0.05104 (17) | 0.72858 (14) | 0.49623 (13) | 0.0425 (3) | |
| C7A | 0.04519 (19) | 0.82716 (16) | 0.40989 (15) | 0.0381 (3) | |
| C8 | 0.1934 (2) | 0.6894 (2) | 0.81865 (16) | 0.0472 (4) | |
| H8A | 0.0833 | 0.6883 | 0.8704 | 0.057* | |
| H8B | 0.2597 | 0.7636 | 0.8442 | 0.057* | |
| C9 | 0.3071 (2) | 0.52937 (18) | 0.85475 (15) | 0.0414 (4) | |
| C10 | 0.2802 (2) | 0.4252 (2) | 0.97390 (17) | 0.0529 (4) | |
| H10 | 0.1897 | 0.4529 | 1.0318 | 0.063* | |
| C11 | 0.3881 (3) | 0.2788 (2) | 1.0076 (2) | 0.0665 (6) | |
| H11 | 0.3689 | 0.2092 | 1.0880 | 0.080* | |
| C12 | 0.5219 (3) | 0.2363 (2) | 0.9239 (2) | 0.0631 (5) | |
| H12 | 0.5924 | 0.1377 | 0.9464 | 0.076* | |
| C13 | 0.5515 (3) | 0.3398 (2) | 0.8065 (2) | 0.0616 (5) | |
| H13 | 0.6436 | 0.3123 | 0.7498 | 0.074* | |
| C14 | 0.4449 (2) | 0.4849 (2) | 0.77241 (18) | 0.0532 (4) | |
| H14 | 0.4660 | 0.5543 | 0.6924 | 0.064* | |
| O15 | 0.36170 (17) | 0.89873 (15) | 0.64121 (13) | 0.0601 (4) | |
| C16 | −0.0948 (3) | 0.8539 (3) | 0.1761 (2) | 0.0701 (6) | |
| H16A | −0.0809 | 0.7434 | 0.1835 | 0.105* | |
| H16B | −0.0629 | 0.9046 | 0.0869 | 0.105* | |
| H16C | −0.2186 | 0.8931 | 0.1922 | 0.105* | |
| C17 | −0.2185 (3) | 0.6832 (2) | 0.4563 (2) | 0.0634 (5) | |
| H17A | −0.3088 | 0.7746 | 0.4285 | 0.095* | |
| H17B | −0.2615 | 0.6128 | 0.5318 | 0.095* | |
| H17C | −0.1947 | 0.6326 | 0.3826 | 0.095* | |
| O18 | −0.07452 (17) | 0.58907 (15) | 0.71081 (13) | 0.0605 (4) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| N1 | 0.0449 (7) | 0.0438 (7) | 0.0448 (7) | −0.0085 (6) | −0.0043 (6) | −0.0051 (6) |
| N2 | 0.0491 (8) | 0.0458 (7) | 0.0509 (8) | −0.0100 (6) | 0.0032 (6) | −0.0011 (6) |
| C3 | 0.0401 (8) | 0.0409 (8) | 0.0549 (9) | −0.0105 (6) | 0.0044 (7) | −0.0056 (7) |
| C3A | 0.0327 (7) | 0.0367 (7) | 0.0485 (8) | −0.0073 (6) | 0.0009 (6) | −0.0084 (6) |
| C4 | 0.0332 (7) | 0.0412 (8) | 0.0523 (9) | −0.0081 (6) | −0.0018 (6) | −0.0107 (7) |
| N5 | 0.0374 (6) | 0.0430 (7) | 0.0426 (7) | −0.0088 (5) | −0.0020 (5) | −0.0073 (5) |
| C6 | 0.0378 (8) | 0.0422 (8) | 0.0476 (9) | −0.0108 (6) | 0.0027 (6) | −0.0079 (7) |
| N7 | 0.0377 (7) | 0.0422 (7) | 0.0492 (7) | −0.0152 (5) | −0.0030 (6) | −0.0074 (6) |
| C7A | 0.0351 (7) | 0.0331 (7) | 0.0453 (8) | −0.0042 (6) | 0.0000 (6) | −0.0082 (6) |
| C8 | 0.0467 (9) | 0.0544 (9) | 0.0409 (8) | −0.0086 (7) | −0.0003 (7) | −0.0119 (7) |
| C9 | 0.0396 (8) | 0.0477 (8) | 0.0391 (8) | −0.0140 (6) | −0.0034 (6) | −0.0092 (6) |
| C10 | 0.0493 (9) | 0.0656 (11) | 0.0435 (9) | −0.0210 (8) | −0.0018 (7) | −0.0036 (8) |
| C11 | 0.0752 (13) | 0.0626 (11) | 0.0540 (11) | −0.0254 (10) | −0.0164 (10) | 0.0129 (9) |
| C12 | 0.0678 (12) | 0.0474 (10) | 0.0698 (12) | −0.0043 (9) | −0.0219 (10) | −0.0065 (9) |
| C13 | 0.0598 (11) | 0.0586 (11) | 0.0635 (11) | 0.0016 (9) | −0.0015 (9) | −0.0159 (9) |
| C14 | 0.0568 (10) | 0.0504 (9) | 0.0482 (9) | −0.0062 (8) | 0.0055 (8) | −0.0050 (7) |
| O15 | 0.0507 (7) | 0.0701 (8) | 0.0639 (8) | −0.0280 (6) | −0.0115 (6) | −0.0110 (6) |
| C16 | 0.0857 (14) | 0.0780 (13) | 0.0489 (10) | −0.0279 (11) | −0.0174 (10) | −0.0077 (9) |
| C17 | 0.0544 (10) | 0.0703 (12) | 0.0692 (12) | −0.0340 (9) | −0.0109 (9) | −0.0062 (9) |
| O18 | 0.0584 (8) | 0.0665 (8) | 0.0564 (7) | −0.0303 (6) | 0.0048 (6) | −0.0002 (6) |
Geometric parameters (Å, º) top
| N1—C7A | 1.343 (2) | C8—H8B | 0.9700 |
| N1—N2 | 1.380 (2) | C9—C10 | 1.380 (2) |
| N1—C16 | 1.457 (2) | C9—C14 | 1.382 (2) |
| N2—C3 | 1.314 (2) | C10—C11 | 1.390 (3) |
| C3—C3A | 1.404 (2) | C10—H10 | 0.9300 |
| C3—H3 | 0.9300 | C11—C12 | 1.366 (3) |
| C3A—C7A | 1.380 (2) | C11—H11 | 0.9300 |
| C3A—C4 | 1.425 (2) | C12—C13 | 1.369 (3) |
| C4—O15 | 1.2211 (19) | C12—H12 | 0.9300 |
| C4—N5 | 1.404 (2) | C13—C14 | 1.378 (3) |
| N5—C6 | 1.392 (2) | C13—H13 | 0.9300 |
| N5—C8 | 1.466 (2) | C14—H14 | 0.9300 |
| C6—O18 | 1.2161 (19) | C16—H16A | 0.9600 |
| C6—N7 | 1.384 (2) | C16—H16B | 0.9600 |
| N7—C7A | 1.3747 (19) | C16—H16C | 0.9600 |
| N7—C17 | 1.467 (2) | C17—H17A | 0.9600 |
| C8—C9 | 1.506 (2) | C17—H17B | 0.9600 |
| C8—H8A | 0.9700 | C17—H17C | 0.9600 |
| | | |
| C7A—N1—N2 | 110.53 (13) | C10—C9—C14 | 118.21 (16) |
| C7A—N1—C16 | 131.66 (15) | C10—C9—C8 | 120.58 (15) |
| N2—N1—C16 | 117.62 (14) | C14—C9—C8 | 121.18 (14) |
| C3—N2—N1 | 105.63 (13) | C9—C10—C11 | 120.16 (17) |
| N2—C3—C3A | 111.42 (14) | C9—C10—H10 | 119.9 |
| N2—C3—H3 | 124.3 | C11—C10—H10 | 119.9 |
| C3A—C3—H3 | 124.3 | C12—C11—C10 | 120.72 (17) |
| C7A—C3A—C3 | 104.76 (14) | C12—C11—H11 | 119.6 |
| C7A—C3A—C4 | 121.32 (14) | C10—C11—H11 | 119.6 |
| C3—C3A—C4 | 133.90 (14) | C11—C12—C13 | 119.51 (18) |
| O15—C4—N5 | 120.59 (15) | C11—C12—H12 | 120.2 |
| O15—C4—C3A | 126.27 (15) | C13—C12—H12 | 120.2 |
| N5—C4—C3A | 113.14 (13) | C12—C13—C14 | 120.06 (19) |
| C6—N5—C4 | 125.95 (13) | C12—C13—H13 | 120.0 |
| C6—N5—C8 | 115.78 (13) | C14—C13—H13 | 120.0 |
| C4—N5—C8 | 118.27 (13) | C13—C14—C9 | 121.32 (17) |
| O18—C6—N7 | 121.19 (14) | C13—C14—H14 | 119.3 |
| O18—C6—N5 | 120.85 (15) | C9—C14—H14 | 119.3 |
| N7—C6—N5 | 117.96 (13) | N1—C16—H16A | 109.5 |
| C7A—N7—C6 | 118.68 (13) | N1—C16—H16B | 109.5 |
| C7A—N7—C17 | 123.55 (14) | H16A—C16—H16B | 109.5 |
| C6—N7—C17 | 117.47 (14) | N1—C16—H16C | 109.5 |
| N1—C7A—N7 | 129.51 (14) | H16A—C16—H16C | 109.5 |
| N1—C7A—C3A | 107.65 (14) | H16B—C16—H16C | 109.5 |
| N7—C7A—C3A | 122.83 (14) | N7—C17—H17A | 109.5 |
| N5—C8—C9 | 112.97 (13) | N7—C17—H17B | 109.5 |
| N5—C8—H8A | 109.0 | H17A—C17—H17B | 109.5 |
| C9—C8—H8A | 109.0 | N7—C17—H17C | 109.5 |
| N5—C8—H8B | 109.0 | H17A—C17—H17C | 109.5 |
| C9—C8—H8B | 109.0 | H17B—C17—H17C | 109.5 |
| H8A—C8—H8B | 107.8 | | |
| | | |
| C7A—N1—N2—C3 | −0.24 (17) | N2—N1—C7A—C3A | −0.12 (17) |
| C16—N1—N2—C3 | −175.89 (15) | C16—N1—C7A—C3A | 174.72 (18) |
| N1—N2—C3—C3A | 0.50 (18) | C6—N7—C7A—N1 | 177.97 (14) |
| N2—C3—C3A—C7A | −0.57 (18) | C17—N7—C7A—N1 | −8.5 (3) |
| N2—C3—C3A—C4 | −179.27 (16) | C6—N7—C7A—C3A | −3.3 (2) |
| C7A—C3A—C4—O15 | −177.37 (15) | C17—N7—C7A—C3A | 170.28 (15) |
| C3—C3A—C4—O15 | 1.2 (3) | C3—C3A—C7A—N1 | 0.40 (16) |
| C7A—C3A—C4—N5 | 2.6 (2) | C4—C3A—C7A—N1 | 179.30 (13) |
| C3—C3A—C4—N5 | −178.90 (16) | C3—C3A—C7A—N7 | −178.60 (13) |
| O15—C4—N5—C6 | 177.15 (14) | C4—C3A—C7A—N7 | 0.3 (2) |
| C3A—C4—N5—C6 | −2.8 (2) | C6—N5—C8—C9 | 79.46 (17) |
| O15—C4—N5—C8 | −1.7 (2) | C4—N5—C8—C9 | −101.57 (16) |
| C3A—C4—N5—C8 | 178.34 (12) | N5—C8—C9—C10 | −141.39 (15) |
| C4—N5—C6—O18 | −179.73 (14) | N5—C8—C9—C14 | 41.0 (2) |
| C8—N5—C6—O18 | −0.9 (2) | C14—C9—C10—C11 | −1.1 (2) |
| C4—N5—C6—N7 | 0.1 (2) | C8—C9—C10—C11 | −178.77 (15) |
| C8—N5—C6—N7 | 178.93 (13) | C9—C10—C11—C12 | 0.2 (3) |
| O18—C6—N7—C7A | −177.18 (14) | C10—C11—C12—C13 | 0.9 (3) |
| N5—C6—N7—C7A | 3.0 (2) | C11—C12—C13—C14 | −1.0 (3) |
| O18—C6—N7—C17 | 8.9 (2) | C12—C13—C14—C9 | 0.1 (3) |
| N5—C6—N7—C17 | −170.91 (14) | C10—C9—C14—C13 | 0.9 (3) |
| N2—N1—C7A—N7 | 178.80 (14) | C8—C9—C14—C13 | 178.64 (16) |
| C16—N1—C7A—N7 | −6.4 (3) | | |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C3—H3···O15i | 0.93 | 2.42 | 3.317 (2) | 161 |
| Symmetry code: (i) −x+1, −y+2, −z+1. |
(II) 1-benzyl-5,7-dimethyl-4-dioxo-4,5,6,7-tetrahydropyrazolo[3,4-
d]pyrimidine
top
Crystal data top
| C14H14N4O2 | Dx = 1.356 Mg m−3 |
| Mr = 270.29 | Melting point: 142 K |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| a = 12.468 (1) Å | Cell parameters from 43 reflections |
| b = 7.449 (1) Å | θ = 5.1–12.5° |
| c = 15.076 (2) Å | µ = 0.10 mm−1 |
| β = 108.94 (1)° | T = 293 K |
| V = 1324.4 (3) Å3 | Block, colourless |
| Z = 4 | 0.38 × 0.28 × 0.20 mm |
| F(000) = 568 | |
Data collection top
Bruker P4
diffractometer | Rint = 0.089 |
| Radiation source: fine-focus sealed tube | θmax = 27.0°, θmin = 1.9° |
| Graphite monochromator | h = −1→15 |
| θ–2θ scans | k = −1→9 |
| 3782 measured reflections | l = −19→18 |
| 2885 independent reflections | 3 standard reflections every 97 reflections |
| 1504 reflections with I > 2σ(I) | intensity decay: none |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.059 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.159 | H-atom parameters constrained |
| S = 1.01 | w = 1/[σ2(Fo2) + (0.0653P)2 + 0.0892P]
where P = (Fo2 + 2Fc2)/3 |
| 2885 reflections | (Δ/σ)max < 0.001 |
| 183 parameters | Δρmax = 0.17 e Å−3 |
| 0 restraints | Δρmin = −0.22 e Å−3 |
Crystal data top
| C14H14N4O2 | V = 1324.4 (3) Å3 |
| Mr = 270.29 | Z = 4 |
| Monoclinic, P21/n | Mo Kα radiation |
| a = 12.468 (1) Å | µ = 0.10 mm−1 |
| b = 7.449 (1) Å | T = 293 K |
| c = 15.076 (2) Å | 0.38 × 0.28 × 0.20 mm |
| β = 108.94 (1)° | |
Data collection top
Bruker P4
diffractometer | Rint = 0.089 |
| 3782 measured reflections | 3 standard reflections every 97 reflections |
| 2885 independent reflections | intensity decay: none |
| 1504 reflections with I > 2σ(I) | |
Refinement top
| R[F2 > 2σ(F2)] = 0.059 | 0 restraints |
| wR(F2) = 0.159 | H-atom parameters constrained |
| S = 1.01 | Δρmax = 0.17 e Å−3 |
| 2885 reflections | Δρmin = −0.22 e Å−3 |
| 183 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| N1 | 0.20916 (16) | 0.9282 (3) | 0.00658 (13) | 0.0581 (6) | |
| N2 | 0.1691 (2) | 0.9101 (3) | −0.08991 (14) | 0.0661 (6) | |
| C3 | 0.0697 (2) | 0.8320 (4) | −0.10896 (17) | 0.0633 (7) | |
| H3 | 0.0223 | 0.8059 | −0.1693 | 0.076* | |
| C3A | 0.0439 (2) | 0.7932 (3) | −0.02766 (15) | 0.0511 (6) | |
| C4 | −0.0514 (2) | 0.7118 (3) | −0.00995 (17) | 0.0560 (6) | |
| N5 | −0.03889 (15) | 0.7023 (3) | 0.08547 (13) | 0.0539 (5) | |
| C6 | 0.0535 (2) | 0.7604 (3) | 0.15857 (16) | 0.0525 (6) | |
| N7 | 0.14192 (15) | 0.8447 (3) | 0.13688 (12) | 0.0513 (5) | |
| C7A | 0.13447 (19) | 0.8556 (3) | 0.04462 (15) | 0.0493 (6) | |
| C8 | 0.3228 (2) | 0.9998 (4) | 0.04671 (19) | 0.0663 (7) | |
| H8A | 0.3236 | 1.0818 | 0.0969 | 0.080* | |
| H8B | 0.3411 | 1.0686 | −0.0011 | 0.080* | |
| C9 | 0.4136 (2) | 0.8603 (4) | 0.08494 (17) | 0.0580 (6) | |
| C10 | 0.3924 (2) | 0.6790 (4) | 0.0765 (2) | 0.0835 (9) | |
| H10 | 0.3190 | 0.6383 | 0.0463 | 0.100* | |
| C11 | 0.4787 (3) | 0.5570 (5) | 0.1123 (3) | 0.1017 (11) | |
| H11 | 0.4634 | 0.4345 | 0.1069 | 0.122* | |
| C12 | 0.5870 (3) | 0.6161 (7) | 0.1558 (2) | 0.0965 (12) | |
| H12 | 0.6458 | 0.5341 | 0.1787 | 0.116* | |
| C13 | 0.6081 (3) | 0.7957 (7) | 0.1656 (2) | 0.0937 (11) | |
| H13 | 0.6813 | 0.8364 | 0.1963 | 0.112* | |
| C14 | 0.5226 (2) | 0.9156 (5) | 0.13061 (19) | 0.0771 (9) | |
| H14 | 0.5383 | 1.0378 | 0.1378 | 0.093* | |
| O15 | −0.13713 (16) | 0.6565 (3) | −0.06912 (12) | 0.0795 (6) | |
| C16 | −0.1335 (2) | 0.6290 (4) | 0.1117 (2) | 0.0709 (8) | |
| H16A | −0.1072 | 0.5297 | 0.1537 | 0.106* | |
| H16B | −0.1920 | 0.5889 | 0.0564 | 0.106* | |
| H16C | −0.1631 | 0.7205 | 0.1421 | 0.106* | |
| O17 | 0.05736 (15) | 0.7429 (3) | 0.23962 (12) | 0.0705 (6) | |
| C18 | 0.2302 (2) | 0.9323 (4) | 0.21321 (17) | 0.0726 (8) | |
| H18A | 0.3026 | 0.8811 | 0.2183 | 0.109* | |
| H18B | 0.2148 | 0.9150 | 0.2710 | 0.109* | |
| H18C | 0.2310 | 1.0585 | 0.2004 | 0.109* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| N1 | 0.0600 (12) | 0.0600 (13) | 0.0538 (12) | 0.0080 (11) | 0.0178 (10) | 0.0070 (11) |
| N2 | 0.0790 (15) | 0.0691 (15) | 0.0534 (12) | 0.0141 (13) | 0.0260 (11) | 0.0080 (11) |
| C3 | 0.0744 (18) | 0.0637 (18) | 0.0489 (14) | 0.0156 (15) | 0.0160 (13) | −0.0008 (13) |
| C3A | 0.0534 (14) | 0.0521 (13) | 0.0428 (12) | 0.0139 (12) | 0.0088 (11) | 0.0003 (11) |
| C4 | 0.0579 (15) | 0.0520 (15) | 0.0519 (14) | 0.0117 (12) | 0.0091 (12) | −0.0065 (12) |
| N5 | 0.0511 (11) | 0.0527 (13) | 0.0563 (12) | 0.0056 (10) | 0.0152 (9) | −0.0015 (10) |
| C6 | 0.0595 (15) | 0.0509 (15) | 0.0462 (13) | 0.0128 (12) | 0.0160 (12) | 0.0006 (11) |
| N7 | 0.0520 (11) | 0.0557 (12) | 0.0419 (10) | 0.0032 (10) | 0.0092 (9) | 0.0012 (9) |
| C7A | 0.0543 (13) | 0.0466 (13) | 0.0468 (12) | 0.0145 (12) | 0.0162 (11) | 0.0064 (11) |
| C8 | 0.0630 (16) | 0.0637 (17) | 0.0756 (17) | −0.0007 (14) | 0.0275 (13) | 0.0081 (14) |
| C9 | 0.0565 (15) | 0.0669 (17) | 0.0568 (14) | 0.0002 (13) | 0.0268 (12) | 0.0021 (13) |
| C10 | 0.0636 (17) | 0.072 (2) | 0.110 (2) | 0.0072 (16) | 0.0207 (16) | 0.0067 (18) |
| C11 | 0.096 (3) | 0.084 (2) | 0.130 (3) | 0.027 (2) | 0.044 (2) | 0.021 (2) |
| C12 | 0.079 (2) | 0.137 (4) | 0.082 (2) | 0.047 (2) | 0.0378 (18) | 0.021 (2) |
| C13 | 0.0597 (19) | 0.143 (4) | 0.078 (2) | 0.012 (2) | 0.0226 (15) | −0.015 (2) |
| C14 | 0.0610 (17) | 0.094 (2) | 0.0831 (19) | −0.0039 (17) | 0.0327 (15) | −0.0126 (17) |
| O15 | 0.0662 (12) | 0.0913 (15) | 0.0668 (12) | −0.0064 (11) | 0.0020 (10) | −0.0121 (11) |
| C16 | 0.0677 (17) | 0.0635 (17) | 0.0863 (19) | −0.0038 (14) | 0.0316 (15) | −0.0029 (15) |
| O17 | 0.0825 (12) | 0.0779 (14) | 0.0543 (11) | −0.0013 (10) | 0.0265 (9) | 0.0033 (9) |
| C18 | 0.0718 (17) | 0.085 (2) | 0.0521 (14) | −0.0112 (15) | 0.0081 (13) | −0.0018 (14) |
Geometric parameters (Å, º) top
| N1—C7A | 1.355 (3) | C8—H8B | 0.9700 |
| N1—N2 | 1.383 (3) | C9—C14 | 1.373 (3) |
| N1—C8 | 1.449 (3) | C9—C10 | 1.374 (4) |
| N2—C3 | 1.314 (3) | C10—C11 | 1.378 (4) |
| C3—C3A | 1.395 (3) | C10—H10 | 0.9300 |
| C3—H3 | 0.9300 | C11—C12 | 1.368 (5) |
| C3A—C7A | 1.371 (3) | C11—H11 | 0.9300 |
| C3A—C4 | 1.434 (4) | C12—C13 | 1.362 (5) |
| C4—O15 | 1.220 (3) | C12—H12 | 0.9300 |
| C4—N5 | 1.398 (3) | C13—C14 | 1.359 (5) |
| N5—C6 | 1.380 (3) | C13—H13 | 0.9300 |
| N5—C16 | 1.465 (3) | C14—H14 | 0.9300 |
| C6—O17 | 1.214 (3) | C16—H16A | 0.9600 |
| C6—N7 | 1.396 (3) | C16—H16B | 0.9600 |
| N7—C7A | 1.366 (3) | C16—H16C | 0.9600 |
| N7—C18 | 1.463 (3) | C18—H18A | 0.9600 |
| C8—C9 | 1.507 (4) | C18—H18B | 0.9600 |
| C8—H8A | 0.9700 | C18—H18C | 0.9600 |
| | | |
| C7A—N1—N2 | 110.08 (19) | C14—C9—C10 | 118.1 (3) |
| C7A—N1—C8 | 133.1 (2) | C14—C9—C8 | 118.9 (3) |
| N2—N1—C8 | 116.4 (2) | C10—C9—C8 | 122.9 (2) |
| C3—N2—N1 | 105.41 (19) | C9—C10—C11 | 120.6 (3) |
| N2—C3—C3A | 111.7 (2) | C9—C10—H10 | 119.7 |
| N2—C3—H3 | 124.1 | C11—C10—H10 | 119.7 |
| C3A—C3—H3 | 124.1 | C12—C11—C10 | 120.0 (4) |
| C7A—C3A—C3 | 105.2 (2) | C12—C11—H11 | 120.0 |
| C7A—C3A—C4 | 120.9 (2) | C10—C11—H11 | 120.0 |
| C3—C3A—C4 | 133.8 (2) | C13—C12—C11 | 119.6 (3) |
| O15—C4—N5 | 120.9 (2) | C13—C12—H12 | 120.2 |
| O15—C4—C3A | 126.1 (2) | C11—C12—H12 | 120.2 |
| N5—C4—C3A | 113.0 (2) | C14—C13—C12 | 120.2 (3) |
| C6—N5—C4 | 126.2 (2) | C14—C13—H13 | 119.9 |
| C6—N5—C16 | 116.0 (2) | C12—C13—H13 | 119.9 |
| C4—N5—C16 | 117.8 (2) | C13—C14—C9 | 121.4 (3) |
| O17—C6—N5 | 121.3 (2) | C13—C14—H14 | 119.3 |
| O17—C6—N7 | 120.5 (2) | C9—C14—H14 | 119.3 |
| N5—C6—N7 | 118.2 (2) | N5—C16—H16A | 109.5 |
| C7A—N7—C6 | 117.86 (19) | N5—C16—H16B | 109.5 |
| C7A—N7—C18 | 124.0 (2) | H16A—C16—H16B | 109.5 |
| C6—N7—C18 | 117.76 (19) | N5—C16—H16C | 109.5 |
| N1—C7A—N7 | 128.7 (2) | H16A—C16—H16C | 109.5 |
| N1—C7A—C3A | 107.5 (2) | H16B—C16—H16C | 109.5 |
| N7—C7A—C3A | 123.7 (2) | N7—C18—H18A | 109.5 |
| N1—C8—C9 | 114.7 (2) | N7—C18—H18B | 109.5 |
| N1—C8—H8A | 108.6 | H18A—C18—H18B | 109.5 |
| C9—C8—H8A | 108.6 | N7—C18—H18C | 109.5 |
| N1—C8—H8B | 108.6 | H18A—C18—H18C | 109.5 |
| C9—C8—H8B | 108.6 | H18B—C18—H18C | 109.5 |
| H8A—C8—H8B | 107.6 | | |
| | | |
| C7A—N1—N2—C3 | 1.9 (3) | N2—N1—C7A—C3A | −1.3 (3) |
| C8—N1—N2—C3 | 175.6 (2) | C8—N1—C7A—C3A | −173.7 (2) |
| N1—N2—C3—C3A | −1.7 (3) | C6—N7—C7A—N1 | −179.2 (2) |
| N2—C3—C3A—C7A | 0.9 (3) | C18—N7—C7A—N1 | 7.8 (4) |
| N2—C3—C3A—C4 | 179.6 (3) | C6—N7—C7A—C3A | 1.9 (3) |
| C7A—C3A—C4—O15 | 177.9 (2) | C18—N7—C7A—C3A | −171.2 (2) |
| C3—C3A—C4—O15 | −0.6 (5) | C3—C3A—C7A—N1 | 0.3 (3) |
| C7A—C3A—C4—N5 | −1.1 (3) | C4—C3A—C7A—N1 | −178.6 (2) |
| C3—C3A—C4—N5 | −179.6 (2) | C3—C3A—C7A—N7 | 179.4 (2) |
| O15—C4—N5—C6 | −179.8 (2) | C4—C3A—C7A—N7 | 0.5 (4) |
| C3A—C4—N5—C6 | −0.8 (3) | C7A—N1—C8—C9 | 73.6 (3) |
| O15—C4—N5—C16 | −1.8 (3) | N2—N1—C8—C9 | −98.4 (2) |
| C3A—C4—N5—C16 | 177.2 (2) | N1—C8—C9—C14 | −175.2 (2) |
| C4—N5—C6—O17 | −178.5 (2) | N1—C8—C9—C10 | 4.6 (4) |
| C16—N5—C6—O17 | 3.5 (3) | C14—C9—C10—C11 | −0.6 (4) |
| C4—N5—C6—N7 | 3.2 (3) | C8—C9—C10—C11 | 179.6 (3) |
| C16—N5—C6—N7 | −174.9 (2) | C9—C10—C11—C12 | −0.7 (5) |
| O17—C6—N7—C7A | 178.1 (2) | C10—C11—C12—C13 | 1.7 (5) |
| N5—C6—N7—C7A | −3.6 (3) | C11—C12—C13—C14 | −1.3 (5) |
| O17—C6—N7—C18 | −8.4 (4) | C12—C13—C14—C9 | 0.0 (5) |
| N5—C6—N7—C18 | 170.0 (2) | C10—C9—C14—C13 | 0.9 (4) |
| N2—N1—C7A—N7 | 179.6 (2) | C8—C9—C14—C13 | −179.3 (3) |
| C8—N1—C7A—N7 | 7.3 (4) | | |
Experimental details
| | (I) | (II) |
| Crystal data |
| Chemical formula | C14H14N4O2 | C14H14N4O2 |
| Mr | 270.29 | 270.29 |
| Crystal system, space group | Triclinic, P1 | Monoclinic, P21/n |
| Temperature (K) | 293 | 293 |
| a, b, c (Å) | 7.476 (1), 8.923 (1), 10.155 (1) | 12.468 (1), 7.449 (1), 15.076 (2) |
| α, β, γ (°) | 76.68 (1), 89.08 (1), 80.66 (1) | 90, 108.94 (1), 90 |
| V (Å3) | 650.3 (1) | 1324.4 (3) |
| Z | 2 | 4 |
| Radiation type | Mo Kα | Mo Kα |
| µ (mm−1) | 0.10 | 0.10 |
| Crystal size (mm) | 0.45 × 0.30 × 0.20 | 0.38 × 0.28 × 0.20 |
| |
| Data collection |
| Diffractometer | Bruker P4
diffractometer | Bruker P4
diffractometer |
| Absorption correction | – | – |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 3155, 2539, 2128 | 3782, 2885, 1504 |
| Rint | 0.015 | 0.089 |
| (sin θ/λ)max (Å−1) | 0.617 | 0.639 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.046, 0.134, 1.05 | 0.059, 0.159, 1.01 |
| No. of reflections | 2539 | 2885 |
| No. of parameters | 183 | 183 |
| H-atom treatment | H-atom parameters constrained | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.16, −0.30 | 0.17, −0.22 |
Hydrogen-bond geometry (Å, º) for (I) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C3—H3···O15i | 0.93 | 2.42 | 3.317 (2) | 161 |
| Symmetry code: (i) −x+1, −y+2, −z+1. |


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2002/06/00/gd1201/gd1201fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2002/06/00/gd1201/gd1201fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2002/06/00/gd1201/gd1201fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/c/issues/2002/06/00/gd1201/gd1201fig4thm.gif)
![[Figure 5]](http://journals.iucr.org/c/issues/2002/06/00/gd1201/gd1201fig5thm.gif)




Xanthine (3,7-dihydro-1H-purine-2,6-thione) compounds are well known for their intermolecular stacking (Falk et al., 1998) and C—H···O interactions (Desiraju & Steiner, 1999). Last year, we reported the X-ray structure of 1,3-bis(8-chlorotheophyllin-7-yl)propane, containing the xanthine skeleton, which also shows intermolecular stacking (Maulik et al., 2001). In this communication, we report the X-ray structures of two isomeric compounds, namely 5-benzyl-1,7-dimethyl-4,5,6,7-tetrahydropyrazolo[3,4-d]pyrimidine-4,6-dione, (I), and 1-benzyl-5,7-dimethyl-4,5,6,7-tetrahydropyrazolo[3,4-d]pyrimidine-4,6-dione, (II). The syntheses of these two compounds have been reported previously (Avasthi et al., 1998) and they are derived from the pyrazolo[3,4-d]pyrimidine ring system; however, structurally they are closer to the xanthine system which is well known for its C—H···O interactions (Desiraju & Steiner, 1999). In xanthine compounds, however, two N atoms flank the CH group, while in compounds (I) and (II), there is only one adjacent N atom.
The conformations of (I) and (II), together with the atom-numbering schemes, are shown in Figs. 1 and 4, respectively. The molecules are isomeric and differ from one another by the interchange of methyl and benzyl groups at positions N1 and C5. The pendent benzyl substituents are out of the planes of the pyrazolo[3,4-d]pyrimidine ring systems [twist angle: 83.2° in (I) and 80.4° in (II)]. The crystal packing of (I) reveals the presence of weak intermolecular C—H···O bonding (Table 1). Interestingly, this hydrogen bonding (C3—H3···O15) leads to the dimerization of the molecules (Fig. 2). The crystal packing (Fig. 3) shows further independent intermolecular stacking between the phenyl rings and the pyrazolo[3,4-d]pyrimidine systems due to π–π interactions. Pairs of phenyl rings (symmetry code: 1 - x, 1 - y, 2 - z) overlap with an interplanar separation of 3.511 (2) Å and a centroid–centroid separation of 3.374 (2) Å in a `parallel displaced' orientation. The face-to-face overlapping of the pyrazolo[3,4-d]pyrimidine ring systems (symmetry code: -x, 2 - y, 1 - z) displays an interplanar separation of 3.276 (2) Å and a centroid–centroid separation of 3.374 (2) Å. Both modes of stacking interactions are common in xanthine compounds (Falk et al., 1998). The crystal packing of (II), on the other hand, shows no such dimerization. Intermolecular stacking, however, is still present (Fig. 5) among pairs of pyrazolo[3,4-d]pyrimidine ring systems [symmetry code: -x, 2 - y, -z; interplanar spacing: 3.303 (3) Å; centroid separation: 3.365 (2) Å], in similar orientations to those found in (I). Thus, the crystal structures of (I) and (II) are stabilized mainly by C—H···O and π–π interactions, and van der Waals forces.