Buy article online - an online subscription or single-article purchase is required to access this article.
The structures of two new sulfate complexes are reported, namely di-μ-sulfato-κ
3O,O′:
O′′-bis{aqua[2,4,6-tris(2-pyridyl)-1,3,5-triazine-κ
3N1,
N2,
N6]cadmium(II)} tetrahydrate, [Cd
2(SO
4)
2(C
16H
12N
6)
2(H
2O)
2]·4H
2O, and di-μ-sulfato-κ
2O:
O′-bis[(2,2′:6′,2′′-terpyridine-κ
3N1,
N1′,
N1′′)zinc(II)] dihydrate, [Cd
2(SO
4)
2(C
15H
11N
3)
2]·2H
2O, the former being the first report of a Cd(tpt) complex [tpt is 2,4,6-tris(2-pyridyl)-1,3,5-triazine]. Both compounds crystallize in the space group
P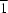
and form centrosymmetric dimeric structures. In the cadmium complex, the metal center is heptacoordinated in the form of a pentagonal bipyramid, while in the zinc complex, the metal ion is in a fivefold environment, the coordination geometry being intermediate between square pyramidal and trigonal bipyramidal. Packing of the dimers leads to the formation of planar structures strongly linked by hydrogen bonding.
Supporting information
CCDC references: 214142; 214143
Both compounds were obtained by direct mixing of an aqueous solution of the cation sulfate (3CdSO4·8H2O or ZnSO4·7H2O), with a methanolic solution of the corresponding organic ligand (tpt or tpy), both in a 0.025 M concentration. A crop of tiny crystals appeared readily, but they were not found adequate for X-ray diffraction and dissolved (those of compound (I) in hot water and those of compound (II) in a 1:1 mixture of hot water and dimethylformamide). On standing, small specimens suitable for the structural study were collected.
H atoms were located at calculated positions, riding on their host atoms. Water H atoms were found in a difference Fourier synthesis and were refined with restrained O—H (0.80 Å) and H···H (> 1.30 Å) distances, and Uiso = 1.2Ueq(O).
For both compounds, data collection: MSC/AFC Diffractometer Control Software (Molecular Structure Corporation, 1988); cell refinement: MSC/AFC Diffractometer Control Software; data reduction: MSC/AFC Diffractometer Control Software; program(s) used to solve structure: SHELXS97 (Sheldrick, 1990); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: XP in SHELXTL/PC (Sheldrick, 1994); software used to prepare material for publication: XP in SHELXTL/PC.
(I) di-µ-sulfato-
κ3O,
O':
O''-bis{aqua[2,4,6-tris(2-pyridyl)-1,3,5-triazine-
κ3N1,
N2,
N6]cadmium(II)} tetrahydrate
top
Crystal data top
| [Cd2(SO4)2(C16H12N6)2(H2O)2]·4H2O | Z = 1 |
| Mr = 1149.68 | F(000) = 576 |
| Triclinic, P1 | Dx = 1.810 Mg m−3 |
| a = 8.870 (2) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 10.640 (2) Å | Cell parameters from 28 reflections |
| c = 12.685 (2) Å | θ = 7.5–15° |
| α = 103.36 (2)° | µ = 1.19 mm−1 |
| β = 98.79 (2)° | T = 293 K |
| γ = 110.45 (2)° | Prism, colorless |
| V = 1055.0 (4) Å3 | 0.20 × 0.12 × 0.05 mm |
Data collection top
Rigaku AFC-7S
diffractometer | 1996 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.039 |
| Graphite monochromator | θmax = 26.0°, θmin = 1.7° |
| ω/2θ scans | h = −5→10 |
Absorption correction: ψ scan
(MSC/AFC Diffractometer Control Software; Molecular Structure Corp, 1988) | k = −13→12 |
| Tmin = 0.83, Tmax = 0.90 | l = −15→13 |
| 5842 measured reflections | 2 standard reflections every 98 reflections |
| 4010 independent reflections | intensity decay: <2% |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.038 | Hydrogen site location: mixed |
| wR(F2) = 0.056 | H atoms treated by a mixture of independent and constrained refinement |
| S = 0.88 | w = 1/[σ2(Fo2))2]
where P = (Fo2 + 2Fc2)/3 |
| 4010 reflections | (Δ/σ)max = 0.003 |
| 319 parameters | Δρmax = 0.44 e Å−3 |
| 13 restraints | Δρmin = −0.40 e Å−3 |
Crystal data top
| [Cd2(SO4)2(C16H12N6)2(H2O)2]·4H2O | γ = 110.45 (2)° |
| Mr = 1149.68 | V = 1055.0 (4) Å3 |
| Triclinic, P1 | Z = 1 |
| a = 8.870 (2) Å | Mo Kα radiation |
| b = 10.640 (2) Å | µ = 1.19 mm−1 |
| c = 12.685 (2) Å | T = 293 K |
| α = 103.36 (2)° | 0.20 × 0.12 × 0.05 mm |
| β = 98.79 (2)° | |
Data collection top
Rigaku AFC-7S
diffractometer | 1996 reflections with I > 2σ(I) |
Absorption correction: ψ scan
(MSC/AFC Diffractometer Control Software; Molecular Structure Corp, 1988) | Rint = 0.039 |
| Tmin = 0.83, Tmax = 0.90 | 2 standard reflections every 98 reflections |
| 5842 measured reflections | intensity decay: <2% |
| 4010 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.038 | 13 restraints |
| wR(F2) = 0.056 | H atoms treated by a mixture of independent and constrained refinement |
| S = 0.88 | Δρmax = 0.44 e Å−3 |
| 4010 reflections | Δρmin = −0.40 e Å−3 |
| 319 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Cd | 0.09400 (5) | −0.00222 (4) | 0.18610 (3) | 0.03625 (13) | |
| S | 0.00314 (17) | −0.20524 (14) | −0.04017 (11) | 0.0375 (4) | |
| O1 | 0.1698 (4) | −0.0991 (3) | 0.0215 (3) | 0.0594 (11) | |
| O2 | −0.1051 (4) | −0.1890 (3) | 0.0335 (3) | 0.0545 (11) | |
| O3 | 0.0025 (4) | −0.3469 (3) | −0.0661 (3) | 0.0571 (11) | |
| O4 | −0.0531 (4) | −0.1834 (3) | −0.1466 (3) | 0.0507 (10) | |
| N1 | 0.3901 (5) | 0.1383 (4) | 0.2518 (3) | 0.0351 (11) | |
| N2 | 0.1686 (5) | 0.1413 (4) | 0.3715 (3) | 0.0326 (11) | |
| N3 | −0.1425 (5) | −0.0291 (4) | 0.2678 (3) | 0.0348 (11) | |
| N4 | 0.3769 (4) | 0.3160 (4) | 0.5268 (3) | 0.0364 (11) | |
| N5 | 0.0946 (5) | 0.2232 (4) | 0.5364 (3) | 0.0331 (11) | |
| N6 | 0.1894 (5) | 0.3838 (4) | 0.7523 (3) | 0.0396 (12) | |
| C1 | 0.4981 (7) | 0.1347 (5) | 0.1910 (4) | 0.0456 (15) | |
| H1A | 0.4584 | 0.0750 | 0.1176 | 0.055* | |
| C2 | 0.6676 (7) | 0.2155 (6) | 0.2314 (4) | 0.0494 (16) | |
| H2A | 0.7397 | 0.2095 | 0.1863 | 0.059* | |
| C3 | 0.7262 (6) | 0.3036 (5) | 0.3381 (5) | 0.0487 (16) | |
| H3A | 0.8395 | 0.3588 | 0.3672 | 0.058* | |
| C4 | 0.6154 (6) | 0.3106 (5) | 0.4037 (4) | 0.0430 (15) | |
| H4A | 0.6528 | 0.3705 | 0.4770 | 0.052* | |
| C5 | 0.4491 (6) | 0.2268 (5) | 0.3574 (4) | 0.0309 (13) | |
| C6 | 0.3253 (6) | 0.2276 (5) | 0.4227 (4) | 0.0308 (13) | |
| C7 | 0.2555 (6) | 0.3077 (5) | 0.5802 (4) | 0.0305 (13) | |
| C8 | 0.0597 (6) | 0.1433 (5) | 0.4310 (4) | 0.0285 (13) | |
| C9 | −0.1169 (6) | 0.0460 (5) | 0.3737 (4) | 0.0303 (13) | |
| C10 | −0.2433 (6) | 0.0343 (5) | 0.4278 (4) | 0.0373 (15) | |
| H10A | −0.2211 | 0.0869 | 0.5022 | 0.045* | |
| C11 | −0.4025 (6) | −0.0575 (5) | 0.3683 (4) | 0.0434 (15) | |
| H11A | −0.4905 | −0.0677 | 0.4018 | 0.052* | |
| C12 | −0.4304 (6) | −0.1341 (5) | 0.2584 (5) | 0.0482 (16) | |
| H12A | −0.5376 | −0.1966 | 0.2171 | 0.058* | |
| C13 | −0.2995 (7) | −0.1179 (5) | 0.2101 (4) | 0.0432 (15) | |
| H13A | −0.3194 | −0.1695 | 0.1357 | 0.052* | |
| C14 | 0.3090 (6) | 0.3974 (5) | 0.6976 (4) | 0.0315 (13) | |
| C15 | 0.4727 (6) | 0.4876 (5) | 0.7468 (4) | 0.0390 (14) | |
| H15A | 0.5528 | 0.4934 | 0.7065 | 0.047* | |
| C16 | 0.5164 (6) | 0.5707 (5) | 0.8589 (4) | 0.0489 (16) | |
| H16A | 0.6260 | 0.6326 | 0.8946 | 0.059* | |
| C17 | 0.3939 (7) | 0.5585 (5) | 0.9145 (4) | 0.0476 (16) | |
| H17A | 0.4184 | 0.6136 | 0.9884 | 0.057* | |
| C18 | 0.2333 (6) | 0.4631 (5) | 0.8591 (4) | 0.0462 (15) | |
| H18A | 0.1515 | 0.4538 | 0.8982 | 0.055* | |
| O1W | 0.1327 (4) | −0.1701 (3) | 0.2716 (3) | 0.0444 (10) | |
| H1WA | 0.056 (4) | −0.204 (4) | 0.301 (4) | 0.053* | |
| H1WB | 0.158 (5) | −0.237 (3) | 0.237 (3) | 0.053* | |
| O2W | 0.8622 (5) | 0.5577 (5) | 0.6671 (3) | 0.0977 (16) | |
| H2WA | 0.898 (4) | 0.536 (3) | 0.727 (2) | 0.117* | |
| H2WB | 0.865 (4) | 0.636 (3) | 0.684 (3) | 0.117* | |
| O3W | 0.1977 (4) | 0.6324 (3) | 0.1193 (3) | 0.0541 (12) | |
| H3WA | 0.142 (5) | 0.541 (3) | 0.109 (4) | 0.065* | |
| H3WB | 0.134 (5) | 0.647 (4) | 0.065 (3) | 0.065* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Cd | 0.0368 (3) | 0.0375 (2) | 0.0315 (2) | 0.01291 (19) | 0.01062 (17) | 0.00717 (18) |
| S | 0.0427 (10) | 0.0338 (9) | 0.0334 (9) | 0.0145 (8) | 0.0095 (7) | 0.0071 (7) |
| O1 | 0.039 (2) | 0.058 (3) | 0.056 (3) | 0.007 (2) | 0.006 (2) | −0.004 (2) |
| O2 | 0.050 (3) | 0.063 (3) | 0.037 (2) | 0.017 (2) | 0.0170 (19) | −0.003 (2) |
| O3 | 0.079 (3) | 0.038 (2) | 0.051 (3) | 0.030 (2) | 0.005 (2) | 0.0043 (19) |
| O4 | 0.077 (3) | 0.048 (2) | 0.031 (2) | 0.031 (2) | 0.0064 (19) | 0.0128 (18) |
| N1 | 0.040 (3) | 0.038 (3) | 0.034 (3) | 0.018 (2) | 0.021 (2) | 0.011 (2) |
| N2 | 0.023 (3) | 0.033 (3) | 0.034 (3) | 0.004 (2) | 0.005 (2) | 0.009 (2) |
| N3 | 0.028 (3) | 0.033 (3) | 0.032 (3) | 0.005 (2) | 0.002 (2) | 0.005 (2) |
| N4 | 0.029 (3) | 0.040 (3) | 0.034 (3) | 0.009 (2) | 0.014 (2) | 0.006 (2) |
| N5 | 0.027 (3) | 0.035 (3) | 0.029 (3) | 0.007 (2) | 0.008 (2) | 0.003 (2) |
| N6 | 0.040 (3) | 0.038 (3) | 0.027 (3) | 0.009 (2) | 0.007 (2) | −0.004 (2) |
| C1 | 0.054 (4) | 0.043 (4) | 0.040 (4) | 0.015 (3) | 0.022 (3) | 0.013 (3) |
| C2 | 0.041 (4) | 0.064 (4) | 0.055 (4) | 0.024 (3) | 0.030 (3) | 0.024 (3) |
| C3 | 0.030 (4) | 0.055 (4) | 0.061 (4) | 0.013 (3) | 0.016 (3) | 0.019 (3) |
| C4 | 0.035 (4) | 0.042 (4) | 0.052 (4) | 0.014 (3) | 0.014 (3) | 0.015 (3) |
| C5 | 0.031 (3) | 0.031 (3) | 0.032 (3) | 0.012 (3) | 0.007 (3) | 0.015 (3) |
| C6 | 0.027 (3) | 0.034 (3) | 0.040 (4) | 0.014 (3) | 0.015 (3) | 0.021 (3) |
| C7 | 0.036 (4) | 0.026 (3) | 0.031 (3) | 0.011 (3) | 0.012 (3) | 0.011 (3) |
| C8 | 0.027 (3) | 0.029 (3) | 0.035 (3) | 0.013 (3) | 0.013 (3) | 0.013 (3) |
| C9 | 0.032 (4) | 0.029 (3) | 0.028 (3) | 0.011 (3) | 0.007 (3) | 0.008 (3) |
| C10 | 0.031 (4) | 0.038 (4) | 0.033 (3) | 0.009 (3) | 0.005 (3) | 0.003 (3) |
| C11 | 0.030 (4) | 0.054 (4) | 0.043 (4) | 0.015 (3) | 0.012 (3) | 0.010 (3) |
| C12 | 0.021 (3) | 0.047 (4) | 0.064 (5) | 0.003 (3) | 0.006 (3) | 0.013 (3) |
| C13 | 0.042 (4) | 0.035 (4) | 0.037 (4) | 0.010 (3) | 0.003 (3) | −0.002 (3) |
| C14 | 0.033 (4) | 0.028 (3) | 0.028 (3) | 0.009 (3) | 0.011 (3) | 0.001 (3) |
| C15 | 0.034 (4) | 0.044 (4) | 0.039 (4) | 0.015 (3) | 0.014 (3) | 0.012 (3) |
| C16 | 0.036 (4) | 0.051 (4) | 0.041 (4) | 0.008 (3) | 0.001 (3) | 0.002 (3) |
| C17 | 0.046 (4) | 0.045 (4) | 0.034 (4) | 0.009 (3) | 0.007 (3) | −0.004 (3) |
| C18 | 0.046 (4) | 0.047 (4) | 0.041 (4) | 0.012 (3) | 0.017 (3) | 0.011 (3) |
| O1W | 0.047 (3) | 0.040 (2) | 0.046 (3) | 0.015 (2) | 0.0190 (18) | 0.013 (2) |
| O2W | 0.084 (3) | 0.122 (4) | 0.056 (3) | 0.016 (3) | 0.019 (2) | 0.009 (3) |
| O3W | 0.050 (3) | 0.041 (2) | 0.057 (3) | 0.015 (2) | 0.000 (2) | 0.004 (2) |
Geometric parameters (Å, º) top
| Cd—O4i | 2.278 (3) | C3—H3A | 0.9300 |
| Cd—N2 | 2.337 (4) | C4—C5 | 1.374 (5) |
| Cd—O2 | 2.354 (3) | C4—H4A | 0.9300 |
| Cd—O1W | 2.387 (4) | C5—C6 | 1.475 (6) |
| Cd—O1 | 2.407 (3) | C7—C14 | 1.471 (6) |
| Cd—N1 | 2.417 (4) | C8—C9 | 1.492 (6) |
| Cd—N3 | 2.437 (4) | C9—C10 | 1.386 (6) |
| S—O3 | 1.465 (3) | C10—C11 | 1.374 (5) |
| S—O2 | 1.463 (3) | C10—H10A | 0.9300 |
| S—O1 | 1.464 (3) | C11—C12 | 1.378 (6) |
| S—O4 | 1.465 (3) | C11—H11A | 0.9300 |
| O4—Cdi | 2.278 (3) | C12—C13 | 1.371 (7) |
| N1—C1 | 1.323 (6) | C12—H12A | 0.9300 |
| N1—C5 | 1.347 (5) | C13—H13A | 0.9300 |
| N2—C8 | 1.316 (5) | C14—C15 | 1.375 (6) |
| N2—C6 | 1.324 (5) | C15—C16 | 1.402 (6) |
| N3—C9 | 1.334 (5) | C15—H15A | 0.9300 |
| N3—C13 | 1.348 (5) | C16—C17 | 1.369 (6) |
| N4—C6 | 1.334 (5) | C16—H16A | 0.9300 |
| N4—C7 | 1.345 (5) | C17—C18 | 1.381 (6) |
| N5—C8 | 1.330 (5) | C17—H17A | 0.9300 |
| N5—C7 | 1.336 (5) | C18—H18A | 0.9300 |
| N6—C18 | 1.334 (5) | O1W—H1WA | 0.84 (2) |
| N6—C14 | 1.339 (6) | O1W—H1WB | 0.86 (2) |
| C1—C2 | 1.386 (6) | O2W—H2WA | 0.89 (2) |
| C1—H1A | 0.9300 | O2W—H2WB | 0.80 (2) |
| C2—C3 | 1.356 (6) | O3W—H3WA | 0.89 (2) |
| C2—H2A | 0.9300 | O3W—H3WB | 0.90 (2) |
| C3—C4 | 1.392 (6) | | |
| | | |
| O4i—Cd—N2 | 83.30 (12) | C5—C4—C3 | 118.3 (5) |
| O4i—Cd—O2 | 100.08 (11) | C5—C4—H4A | 120.8 |
| N2—Cd—O2 | 149.95 (13) | C3—C4—H4A | 120.8 |
| O4i—Cd—O1W | 166.07 (12) | N1—C5—C4 | 122.6 (5) |
| N2—Cd—O1W | 83.48 (13) | N1—C5—C6 | 116.2 (5) |
| O2—Cd—O1W | 89.24 (12) | C4—C5—C6 | 121.1 (5) |
| O4i—Cd—O1 | 101.15 (12) | N2—C6—N4 | 124.7 (5) |
| N2—Cd—O1 | 150.50 (12) | N2—C6—C5 | 116.7 (5) |
| O2—Cd—O1 | 58.61 (11) | N4—C6—C5 | 118.6 (4) |
| O1W—Cd—O1 | 92.53 (12) | N5—C7—N4 | 125.5 (5) |
| O4i—Cd—N1 | 90.00 (12) | N5—C7—C14 | 118.6 (5) |
| N2—Cd—N1 | 68.18 (14) | N4—C7—C14 | 115.9 (5) |
| O2—Cd—N1 | 141.05 (12) | N2—C8—N5 | 125.6 (4) |
| O1W—Cd—N1 | 89.15 (12) | N2—C8—C9 | 116.0 (5) |
| O1—Cd—N1 | 82.59 (12) | N5—C8—C9 | 118.4 (5) |
| O4i—Cd—N3 | 85.05 (12) | N3—C9—C10 | 123.0 (5) |
| N2—Cd—N3 | 66.86 (14) | N3—C9—C8 | 114.9 (5) |
| O2—Cd—N3 | 83.59 (12) | C10—C9—C8 | 122.0 (5) |
| O1W—Cd—N3 | 85.74 (12) | C11—C10—C9 | 118.0 (5) |
| O1—Cd—N3 | 142.20 (11) | C11—C10—H10A | 121.0 |
| N1—Cd—N3 | 135.04 (13) | C9—C10—H10A | 121.0 |
| O3—S—O2 | 111.2 (2) | C10—C11—C12 | 119.3 (5) |
| O3—S—O1 | 110.57 (19) | C10—C11—H11A | 120.4 |
| O2—S—O1 | 105.5 (2) | C12—C11—H11A | 120.4 |
| O3—S—O4 | 107.48 (19) | C13—C12—C11 | 119.7 (5) |
| O2—S—O4 | 110.7 (2) | C13—C12—H12A | 120.1 |
| O1—S—O4 | 111.3 (2) | C11—C12—H12A | 120.1 |
| S—O1—Cd | 96.77 (17) | N3—C13—C12 | 121.6 (5) |
| S—O2—Cd | 99.08 (17) | N3—C13—H13A | 119.2 |
| S—O4—Cdi | 131.70 (19) | C12—C13—H13A | 119.2 |
| C1—N1—C5 | 117.8 (5) | N6—C14—C15 | 123.0 (5) |
| C1—N1—Cd | 124.5 (4) | N6—C14—C7 | 115.8 (5) |
| C5—N1—Cd | 117.7 (3) | C15—C14—C7 | 121.2 (5) |
| C8—N2—C6 | 115.9 (5) | C14—C15—C16 | 118.7 (5) |
| C8—N2—Cd | 122.9 (3) | C14—C15—H15A | 120.7 |
| C6—N2—Cd | 121.2 (4) | C16—C15—H15A | 120.7 |
| C9—N3—C13 | 118.3 (5) | C17—C16—C15 | 118.4 (5) |
| C9—N3—Cd | 119.2 (3) | C17—C16—H16A | 120.8 |
| C13—N3—Cd | 122.5 (3) | C15—C16—H16A | 120.8 |
| C6—N4—C7 | 114.2 (4) | C16—C17—C18 | 118.9 (5) |
| C8—N5—C7 | 114.0 (4) | C16—C17—H17A | 120.5 |
| C18—N6—C14 | 117.6 (4) | C18—C17—H17A | 120.5 |
| N1—C1—C2 | 123.2 (5) | N6—C18—C17 | 123.4 (5) |
| N1—C1—H1A | 118.4 | N6—C18—H18A | 118.3 |
| C2—C1—H1A | 118.4 | C17—C18—H18A | 118.3 |
| C3—C2—C1 | 118.7 (5) | Cd—O1W—H1WA | 113 (3) |
| C3—C2—H2A | 120.7 | Cd—O1W—H1WB | 121 (3) |
| C1—C2—H2A | 120.7 | H1WA—O1W—H1WB | 109 (2) |
| C2—C3—C4 | 119.4 (5) | H2WA—O2W—H2WB | 112 (3) |
| C2—C3—H3A | 120.3 | H3WA—O3W—H3WB | 101 (2) |
| C4—C3—H3A | 120.3 | | |
| | | |
| O3—S—O1—Cd | −120.61 (18) | Cd—N1—C1—C2 | −179.3 (4) |
| O2—S—O1—Cd | −0.2 (2) | N1—C1—C2—C3 | −0.5 (8) |
| O4—S—O1—Cd | 119.97 (18) | C1—C2—C3—C4 | −0.1 (8) |
| O4i—Cd—O1—S | −94.93 (17) | C2—C3—C4—C5 | 0.2 (8) |
| N2—Cd—O1—S | 168.9 (2) | C1—N1—C5—C4 | −0.9 (7) |
| O2—Cd—O1—S | 0.16 (15) | Cd—N1—C5—C4 | 179.4 (3) |
| O1W—Cd—O1—S | 87.72 (18) | C1—N1—C5—C6 | −179.8 (4) |
| N1—Cd—O1—S | 176.54 (19) | Cd—N1—C5—C6 | 0.6 (5) |
| N3—Cd—O1—S | 1.4 (3) | C3—C4—C5—N1 | 0.3 (7) |
| O3—S—O2—Cd | 120.19 (17) | C3—C4—C5—C6 | 179.1 (5) |
| O1—S—O2—Cd | 0.2 (2) | C8—N2—C6—N4 | −1.7 (7) |
| O4—S—O2—Cd | −120.37 (18) | Cd—N2—C6—N4 | 177.6 (3) |
| O4i—Cd—O2—S | 96.83 (19) | C8—N2—C6—C5 | 180.0 (4) |
| N2—Cd—O2—S | −169.1 (2) | Cd—N2—C6—C5 | −0.6 (6) |
| O1W—Cd—O2—S | −93.59 (18) | C7—N4—C6—N2 | 2.6 (7) |
| O1—Cd—O2—S | −0.16 (15) | C7—N4—C6—C5 | −179.2 (4) |
| N1—Cd—O2—S | −5.9 (3) | N1—C5—C6—N2 | 0.0 (7) |
| N3—Cd—O2—S | −179.4 (2) | C4—C5—C6—N2 | −178.8 (4) |
| O3—S—O4—Cdi | 178.8 (2) | N1—C5—C6—N4 | −178.4 (4) |
| O2—S—O4—Cdi | 57.2 (3) | C4—C5—C6—N4 | 2.8 (7) |
| O1—S—O4—Cdi | −59.9 (3) | C8—N5—C7—N4 | 0.1 (7) |
| O4i—Cd—N1—C1 | −97.5 (4) | C8—N5—C7—C14 | −178.0 (4) |
| N2—Cd—N1—C1 | 179.7 (4) | C6—N4—C7—N5 | −1.7 (7) |
| O2—Cd—N1—C1 | 8.7 (5) | C6—N4—C7—C14 | 176.4 (4) |
| O1W—Cd—N1—C1 | 96.4 (4) | C6—N2—C8—N5 | −0.2 (7) |
| O1—Cd—N1—C1 | 3.7 (4) | Cd—N2—C8—N5 | −179.6 (3) |
| N3—Cd—N1—C1 | 179.5 (4) | C6—N2—C8—C9 | −179.8 (4) |
| O4i—Cd—N1—C5 | 82.2 (3) | Cd—N2—C8—C9 | 0.8 (6) |
| N2—Cd—N1—C5 | −0.6 (3) | C7—N5—C8—N2 | 0.9 (7) |
| O2—Cd—N1—C5 | −171.7 (3) | C7—N5—C8—C9 | −179.4 (4) |
| O1W—Cd—N1—C5 | −83.9 (3) | C13—N3—C9—C10 | 1.7 (7) |
| O1—Cd—N1—C5 | −176.6 (3) | Cd—N3—C9—C10 | −178.1 (4) |
| N3—Cd—N1—C5 | −0.8 (4) | C13—N3—C9—C8 | −179.1 (4) |
| O4i—Cd—N2—C8 | 87.3 (4) | Cd—N3—C9—C8 | 1.2 (5) |
| O2—Cd—N2—C8 | −11.3 (5) | N2—C8—C9—N3 | −1.3 (6) |
| O1W—Cd—N2—C8 | −88.3 (4) | N5—C8—C9—N3 | 179.0 (4) |
| O1—Cd—N2—C8 | −171.9 (3) | N2—C8—C9—C10 | 178.0 (5) |
| N1—Cd—N2—C8 | 180.0 (4) | N5—C8—C9—C10 | −1.7 (7) |
| N3—Cd—N2—C8 | −0.2 (4) | N3—C9—C10—C11 | −1.2 (8) |
| O4i—Cd—N2—C6 | −92.0 (4) | C8—C9—C10—C11 | 179.6 (4) |
| O2—Cd—N2—C6 | 169.4 (3) | C9—C10—C11—C12 | 0.3 (8) |
| O1W—Cd—N2—C6 | 92.4 (4) | C10—C11—C12—C13 | 0.0 (8) |
| O1—Cd—N2—C6 | 8.8 (5) | C9—N3—C13—C12 | −1.3 (7) |
| N1—Cd—N2—C6 | 0.7 (3) | Cd—N3—C13—C12 | 178.4 (4) |
| N3—Cd—N2—C6 | −179.5 (4) | C11—C12—C13—N3 | 0.5 (8) |
| O4i—Cd—N3—C9 | −85.4 (3) | C18—N6—C14—C15 | 0.5 (8) |
| N2—Cd—N3—C9 | −0.6 (3) | C18—N6—C14—C7 | 179.8 (4) |
| O2—Cd—N3—C9 | 173.8 (3) | N5—C7—C14—N6 | 1.5 (7) |
| O1W—Cd—N3—C9 | 84.1 (3) | N4—C7—C14—N6 | −176.8 (4) |
| O1—Cd—N3—C9 | 172.7 (3) | N5—C7—C14—C15 | −179.2 (4) |
| N1—Cd—N3—C9 | −0.4 (4) | N4—C7—C14—C15 | 2.5 (7) |
| O4i—Cd—N3—C13 | 94.8 (4) | N6—C14—C15—C16 | −0.8 (8) |
| N2—Cd—N3—C13 | 179.7 (4) | C7—C14—C15—C16 | 179.9 (4) |
| O2—Cd—N3—C13 | −5.9 (4) | C14—C15—C16—C17 | −0.1 (8) |
| O1W—Cd—N3—C13 | −95.6 (4) | C15—C16—C17—C18 | 1.3 (8) |
| O1—Cd—N3—C13 | −7.0 (5) | C14—N6—C18—C17 | 0.8 (8) |
| N1—Cd—N3—C13 | 179.9 (3) | C16—C17—C18—N6 | −1.8 (8) |
| C5—N1—C1—C2 | 1.0 (8) | | |
| Symmetry code: (i) −x, −y, −z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O3W—H3WA···O3i | 0.89 (2) | 1.90 (2) | 2.784 (5) | 170 (4) |
| O3W—H3WB···O3ii | 0.90 (2) | 1.91 (2) | 2.800 (4) | 171 (4) |
| O2W—H2WB···O4iii | 0.80 (2) | 2.34 (3) | 2.956 (5) | 134 (3) |
| O2W—H2WA···O3iii | 0.89 (2) | 2.50 (3) | 3.206 (5) | 137 (3) |
| O1W—H1WA···N6iv | 0.84 (2) | 2.21 (3) | 2.881 (5) | 136 (4) |
| O1W—H1WB···O3Wv | 0.86 (2) | 1.96 (3) | 2.791 (5) | 162 (4) |
| Symmetry codes: (i) −x, −y, −z; (ii) x, y+1, z; (iii) x+1, y+1, z+1; (iv) −x, −y, −z+1; (v) x, y−1, z. |
(II) di-µ-sulfato-
κ2O:
O'-bis[(2,2':6',2''-terpyridine-
κ3N1,
N1',
N1'')zinc(II)] dihydrate
top
Crystal data top
| [Cd2(SO4)2(C15H11N3)2]·2H2O | Z = 1 |
| Mr = 825.43 | F(000) = 420 |
| Triclinic, P1 | Dx = 1.800 Mg m−3 |
| a = 9.700 (2) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 9.755 (2) Å | Cell parameters from 30 reflections |
| c = 10.324 (2) Å | θ = 7.5–15° |
| α = 110.34 (2)° | µ = 1.79 mm−1 |
| β = 116.59 (2)° | T = 293 K |
| γ = 98.83 (2)° | Prism, colorless |
| V = 761.4 (4) Å3 | 0.24 × 0.16 × 0.10 mm |
Data collection top
Rigaku AFC-7S
diffractometer | 2008 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.039 |
| Graphite monochromator | θmax = 26.0°, θmin = 2.4° |
| ω/2θ scans | h = −11→11 |
Absorption correction: ψ scan
(MSC/AFC Diffractometer Control Software; Molecular Structure Corp, 1988) | k = −12→4 |
| Tmin = 0.79, Tmax = 0.86 | l = −11→12 |
| 4216 measured reflections | 2 standard reflections every 98 reflections |
| 2890 independent reflections | intensity decay: <2% |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.040 | Hydrogen site location: mixed |
| wR(F2) = 0.073 | H atoms treated by a mixture of independent and constrained refinement |
| S = 0.85 | w = 1/[σ2(Fo2) + (0.0178P)2]
where P = (Fo2 + 2Fc2)/3 |
| 2890 reflections | (Δ/σ)max = 0.004 |
| 234 parameters | Δρmax = 0.41 e Å−3 |
| 3 restraints | Δρmin = −0.35 e Å−3 |
Crystal data top
| [Cd2(SO4)2(C15H11N3)2]·2H2O | γ = 98.83 (2)° |
| Mr = 825.43 | V = 761.4 (4) Å3 |
| Triclinic, P1 | Z = 1 |
| a = 9.700 (2) Å | Mo Kα radiation |
| b = 9.755 (2) Å | µ = 1.79 mm−1 |
| c = 10.324 (2) Å | T = 293 K |
| α = 110.34 (2)° | 0.24 × 0.16 × 0.10 mm |
| β = 116.59 (2)° | |
Data collection top
Rigaku AFC-7S
diffractometer | 2008 reflections with I > 2σ(I) |
Absorption correction: ψ scan
(MSC/AFC Diffractometer Control Software; Molecular Structure Corp, 1988) | Rint = 0.039 |
| Tmin = 0.79, Tmax = 0.86 | 2 standard reflections every 98 reflections |
| 4216 measured reflections | intensity decay: <2% |
| 2890 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.040 | 3 restraints |
| wR(F2) = 0.073 | H atoms treated by a mixture of independent and constrained refinement |
| S = 0.85 | Δρmax = 0.41 e Å−3 |
| 2890 reflections | Δρmin = −0.35 e Å−3 |
| 234 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Zn | 0.76317 (5) | 0.85637 (5) | 0.90932 (5) | 0.03576 (15) | |
| S | 1.07896 (11) | 1.02598 (11) | 1.26397 (12) | 0.0368 (2) | |
| O1 | 0.9204 (3) | 0.8918 (3) | 1.1321 (3) | 0.0613 (8) | |
| O2 | 1.0570 (3) | 1.1164 (3) | 1.3913 (3) | 0.0640 (8) | |
| O3 | 1.2096 (3) | 0.9697 (3) | 1.3206 (4) | 0.0722 (9) | |
| O4 | 1.1123 (3) | 1.1279 (3) | 1.1934 (3) | 0.0431 (7) | |
| N1 | 0.7047 (3) | 0.6013 (3) | 0.8129 (4) | 0.0379 (8) | |
| N2 | 0.6833 (3) | 1.0545 (4) | 0.9480 (4) | 0.0388 (8) | |
| N3 | 0.5075 (3) | 0.7529 (3) | 0.7496 (3) | 0.0340 (7) | |
| C1 | 0.8151 (5) | 0.5312 (5) | 0.8482 (5) | 0.0472 (11) | |
| H1A | 0.9260 | 0.5939 | 0.9286 | 0.057* | |
| C2 | 0.7722 (5) | 0.3712 (5) | 0.7711 (5) | 0.0497 (11) | |
| H2A | 0.8521 | 0.3264 | 0.7978 | 0.060* | |
| C3 | 0.6085 (5) | 0.2790 (5) | 0.6538 (5) | 0.0488 (11) | |
| H3A | 0.5760 | 0.1702 | 0.6002 | 0.059* | |
| C4 | 0.4921 (5) | 0.3477 (4) | 0.6155 (5) | 0.0437 (10) | |
| H4A | 0.3804 | 0.2866 | 0.5372 | 0.052* | |
| C5 | 0.5458 (4) | 0.5102 (4) | 0.6965 (4) | 0.0334 (9) | |
| C6 | 0.4318 (4) | 0.5967 (4) | 0.6603 (4) | 0.0338 (9) | |
| C7 | 0.2630 (4) | 0.5270 (5) | 0.5452 (4) | 0.0442 (10) | |
| H7A | 0.2107 | 0.4181 | 0.4844 | 0.053* | |
| C8 | 0.1744 (5) | 0.6239 (5) | 0.5230 (5) | 0.0521 (11) | |
| H8A | 0.0612 | 0.5802 | 0.4440 | 0.063* | |
| C9 | 0.2521 (4) | 0.7834 (5) | 0.6163 (5) | 0.0486 (11) | |
| H9A | 0.1918 | 0.8483 | 0.6023 | 0.058* | |
| C10 | 0.4199 (4) | 0.8476 (5) | 0.7310 (5) | 0.0394 (9) | |
| C11 | 0.5197 (5) | 1.0156 (5) | 0.8442 (5) | 0.0396 (9) | |
| C12 | 0.4499 (5) | 1.1292 (5) | 0.8477 (5) | 0.0520 (11) | |
| H12A | 0.3371 | 1.1020 | 0.7756 | 0.062* | |
| C13 | 0.5517 (6) | 1.2840 (5) | 0.9610 (5) | 0.0565 (12) | |
| H13A | 0.5069 | 1.3620 | 0.9653 | 0.068* | |
| C14 | 0.7174 (5) | 1.3244 (5) | 1.0669 (5) | 0.0541 (11) | |
| H14A | 0.7865 | 1.4284 | 1.1440 | 0.065* | |
| C15 | 0.7781 (5) | 1.2044 (5) | 1.0550 (5) | 0.0487 (11) | |
| H15A | 0.8909 | 1.2300 | 1.1254 | 0.058* | |
| O1W | 0.8391 (4) | 0.7034 (3) | 0.2766 (4) | 0.0644 (10) | |
| H1WA | 0.858 (5) | 0.757 (4) | 0.367 (4) | 0.077* | |
| H1WB | 0.879 (5) | 0.757 (4) | 0.249 (5) | 0.077* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Zn | 0.0322 (2) | 0.0287 (3) | 0.0355 (3) | 0.0065 (2) | 0.0160 (2) | 0.0095 (2) |
| S | 0.0342 (5) | 0.0292 (6) | 0.0348 (5) | 0.0071 (5) | 0.0155 (4) | 0.0091 (5) |
| O1 | 0.0527 (16) | 0.0488 (19) | 0.0361 (16) | −0.0186 (15) | 0.0050 (14) | 0.0154 (15) |
| O2 | 0.081 (2) | 0.059 (2) | 0.0511 (18) | 0.0186 (18) | 0.0463 (17) | 0.0136 (17) |
| O3 | 0.0602 (19) | 0.062 (2) | 0.104 (3) | 0.0366 (18) | 0.0391 (19) | 0.049 (2) |
| O4 | 0.0459 (15) | 0.0377 (16) | 0.0613 (18) | 0.0169 (14) | 0.0375 (14) | 0.0267 (15) |
| N1 | 0.0332 (17) | 0.0287 (19) | 0.0353 (18) | 0.0036 (15) | 0.0110 (15) | 0.0122 (16) |
| N2 | 0.0385 (18) | 0.0292 (19) | 0.045 (2) | 0.0117 (16) | 0.0248 (16) | 0.0120 (17) |
| N3 | 0.0322 (17) | 0.0309 (18) | 0.0364 (18) | 0.0084 (15) | 0.0197 (15) | 0.0132 (16) |
| C1 | 0.041 (2) | 0.047 (3) | 0.044 (2) | 0.016 (2) | 0.015 (2) | 0.022 (2) |
| C2 | 0.061 (3) | 0.044 (3) | 0.051 (3) | 0.024 (2) | 0.030 (2) | 0.027 (2) |
| C3 | 0.070 (3) | 0.028 (2) | 0.049 (3) | 0.016 (2) | 0.036 (2) | 0.015 (2) |
| C4 | 0.043 (2) | 0.031 (2) | 0.042 (2) | 0.002 (2) | 0.020 (2) | 0.011 (2) |
| C5 | 0.036 (2) | 0.028 (2) | 0.030 (2) | 0.0047 (18) | 0.0186 (18) | 0.0085 (18) |
| C6 | 0.035 (2) | 0.032 (2) | 0.031 (2) | 0.0081 (19) | 0.0188 (18) | 0.0126 (19) |
| C7 | 0.034 (2) | 0.040 (3) | 0.042 (2) | 0.001 (2) | 0.016 (2) | 0.014 (2) |
| C8 | 0.030 (2) | 0.062 (3) | 0.048 (3) | 0.010 (2) | 0.015 (2) | 0.022 (3) |
| C9 | 0.037 (2) | 0.056 (3) | 0.056 (3) | 0.022 (2) | 0.025 (2) | 0.028 (3) |
| C10 | 0.039 (2) | 0.045 (3) | 0.044 (2) | 0.018 (2) | 0.027 (2) | 0.023 (2) |
| C11 | 0.047 (2) | 0.036 (2) | 0.043 (2) | 0.019 (2) | 0.030 (2) | 0.016 (2) |
| C12 | 0.058 (3) | 0.051 (3) | 0.064 (3) | 0.030 (3) | 0.040 (2) | 0.030 (3) |
| C13 | 0.090 (4) | 0.046 (3) | 0.066 (3) | 0.043 (3) | 0.055 (3) | 0.034 (3) |
| C14 | 0.073 (3) | 0.034 (3) | 0.055 (3) | 0.019 (3) | 0.039 (3) | 0.015 (2) |
| C15 | 0.054 (3) | 0.035 (3) | 0.049 (3) | 0.011 (2) | 0.031 (2) | 0.011 (2) |
| O1W | 0.087 (2) | 0.041 (2) | 0.061 (2) | 0.0178 (19) | 0.041 (2) | 0.0203 (17) |
Geometric parameters (Å, º) top
| Zn—O4i | 1.952 (2) | C4—C5 | 1.385 (5) |
| Zn—O1 | 1.970 (3) | C4—H4A | 0.9300 |
| Zn—N3 | 2.074 (3) | C5—C6 | 1.487 (4) |
| Zn—N2 | 2.166 (3) | C6—C7 | 1.382 (4) |
| Zn—N1 | 2.201 (3) | C7—C8 | 1.381 (5) |
| S—O3 | 1.422 (3) | C7—H7A | 0.9300 |
| S—O2 | 1.434 (3) | C8—C9 | 1.366 (5) |
| S—O1 | 1.483 (3) | C8—H8A | 0.9300 |
| S—O4 | 1.496 (2) | C9—C10 | 1.375 (5) |
| O4—Zni | 1.952 (2) | C9—H9A | 0.9300 |
| N1—C5 | 1.337 (4) | C10—C11 | 1.469 (5) |
| N1—C1 | 1.340 (4) | C11—C12 | 1.383 (5) |
| N2—C15 | 1.327 (4) | C12—C13 | 1.379 (5) |
| N2—C11 | 1.350 (4) | C12—H12A | 0.9300 |
| N3—C6 | 1.336 (4) | C13—C14 | 1.367 (5) |
| N3—C10 | 1.353 (4) | C13—H13A | 0.9300 |
| C1—C2 | 1.373 (5) | C14—C15 | 1.381 (5) |
| C1—H1A | 0.9300 | C14—H14A | 0.9300 |
| C2—C3 | 1.371 (5) | C15—H15A | 0.9300 |
| C2—H2A | 0.9300 | O1W—H1WA | 0.81 (3) |
| C3—C4 | 1.377 (5) | O1W—H1WB | 0.81 (3) |
| C3—H3A | 0.9300 | | |
| | | |
| O4i—Zn—O1 | 107.54 (10) | C3—C4—H4A | 120.9 |
| O4i—Zn—N3 | 115.24 (11) | C5—C4—H4A | 120.9 |
| O1—Zn—N3 | 134.66 (11) | N1—C5—C4 | 122.4 (3) |
| O4i—Zn—N2 | 107.83 (10) | N1—C5—C6 | 114.9 (3) |
| O1—Zn—N2 | 104.58 (12) | C4—C5—C6 | 122.7 (3) |
| N3—Zn—N2 | 76.52 (12) | N3—C6—C7 | 121.3 (4) |
| O4i—Zn—N1 | 91.75 (10) | N3—C6—C5 | 113.7 (3) |
| O1—Zn—N1 | 90.39 (11) | C7—C6—C5 | 124.9 (3) |
| N3—Zn—N1 | 74.44 (11) | C8—C7—C6 | 118.0 (4) |
| N2—Zn—N1 | 149.88 (11) | C8—C7—H7A | 121.0 |
| O3—S—O2 | 112.92 (19) | C6—C7—H7A | 121.0 |
| O3—S—O1 | 109.95 (17) | C9—C8—C7 | 120.4 (4) |
| O2—S—O1 | 108.10 (17) | C9—C8—H8A | 119.8 |
| O3—S—O4 | 110.37 (16) | C7—C8—H8A | 119.8 |
| O2—S—O4 | 107.69 (16) | C8—C9—C10 | 119.6 (4) |
| O1—S—O4 | 107.63 (15) | C8—C9—H9A | 120.2 |
| S—O1—Zn | 128.49 (16) | C10—C9—H9A | 120.2 |
| S—O4—Zni | 135.32 (15) | N3—C10—C9 | 120.0 (4) |
| C5—N1—C1 | 118.1 (3) | N3—C10—C11 | 113.9 (3) |
| C5—N1—Zn | 115.7 (2) | C9—C10—C11 | 126.1 (4) |
| C1—N1—Zn | 125.9 (2) | N2—C11—C12 | 121.1 (4) |
| C15—N2—C11 | 119.1 (3) | N2—C11—C10 | 116.8 (3) |
| C15—N2—Zn | 127.0 (3) | C12—C11—C10 | 122.0 (4) |
| C11—N2—Zn | 113.8 (3) | C13—C12—C11 | 118.3 (4) |
| C6—N3—C10 | 120.6 (3) | C13—C12—H12A | 120.8 |
| C6—N3—Zn | 120.9 (2) | C11—C12—H12A | 120.8 |
| C10—N3—Zn | 118.4 (2) | C14—C13—C12 | 120.9 (4) |
| N1—C1—C2 | 123.0 (4) | C14—C13—H13A | 119.5 |
| N1—C1—H1A | 118.5 | C12—C13—H13A | 119.5 |
| C2—C1—H1A | 118.5 | C13—C14—C15 | 117.3 (4) |
| C3—C2—C1 | 118.4 (4) | C13—C14—H14A | 121.3 |
| C3—C2—H2A | 120.8 | C15—C14—H14A | 121.3 |
| C1—C2—H2A | 120.8 | N2—C15—C14 | 123.1 (4) |
| C2—C3—C4 | 119.9 (4) | N2—C15—H15A | 118.4 |
| C2—C3—H3A | 120.0 | C14—C15—H15A | 118.4 |
| C4—C3—H3A | 120.0 | H1WA—O1W—H1WB | 110 (3) |
| C3—C4—C5 | 118.2 (4) | | |
| | | |
| O3—S—O1—Zn | −126.3 (2) | C1—N1—C5—C4 | 1.6 (5) |
| O2—S—O1—Zn | 110.1 (2) | Zn—N1—C5—C4 | 175.1 (3) |
| O4—S—O1—Zn | −6.0 (3) | C1—N1—C5—C6 | −178.0 (3) |
| O4i—Zn—O1—S | 53.6 (2) | Zn—N1—C5—C6 | −4.5 (4) |
| N3—Zn—O1—S | −146.17 (19) | C3—C4—C5—N1 | −1.9 (5) |
| N2—Zn—O1—S | −60.8 (2) | C3—C4—C5—C6 | 177.7 (3) |
| N1—Zn—O1—S | 145.6 (2) | C10—N3—C6—C7 | 0.8 (5) |
| O3—S—O4—Zni | 26.6 (3) | Zn—N3—C6—C7 | −175.4 (2) |
| O2—S—O4—Zni | 150.3 (2) | C10—N3—C6—C5 | −179.7 (3) |
| O1—S—O4—Zni | −93.4 (2) | Zn—N3—C6—C5 | 4.1 (4) |
| O4i—Zn—N1—C5 | −110.8 (2) | N1—C5—C6—N3 | 0.6 (4) |
| O1—Zn—N1—C5 | 141.7 (3) | C4—C5—C6—N3 | −179.1 (3) |
| N3—Zn—N1—C5 | 5.0 (2) | N1—C5—C6—C7 | −180.0 (3) |
| N2—Zn—N1—C5 | 20.8 (4) | C4—C5—C6—C7 | 0.3 (5) |
| O4i—Zn—N1—C1 | 62.1 (3) | N3—C6—C7—C8 | 1.0 (5) |
| O1—Zn—N1—C1 | −45.4 (3) | C5—C6—C7—C8 | −178.4 (3) |
| N3—Zn—N1—C1 | 177.9 (3) | C6—C7—C8—C9 | −1.9 (6) |
| N2—Zn—N1—C1 | −166.3 (3) | C7—C8—C9—C10 | 1.0 (6) |
| O4i—Zn—N2—C15 | −70.2 (3) | C6—N3—C10—C9 | −1.7 (5) |
| O1—Zn—N2—C15 | 44.1 (3) | Zn—N3—C10—C9 | 174.6 (3) |
| N3—Zn—N2—C15 | 177.3 (3) | C6—N3—C10—C11 | 176.9 (3) |
| N1—Zn—N2—C15 | 161.6 (3) | Zn—N3—C10—C11 | −6.8 (4) |
| O4i—Zn—N2—C11 | 106.7 (3) | C8—C9—C10—N3 | 0.7 (6) |
| O1—Zn—N2—C11 | −139.0 (2) | C8—C9—C10—C11 | −177.7 (4) |
| N3—Zn—N2—C11 | −5.8 (2) | C15—N2—C11—C12 | 0.0 (5) |
| N1—Zn—N2—C11 | −21.5 (4) | Zn—N2—C11—C12 | −177.2 (3) |
| O4i—Zn—N3—C6 | 79.6 (3) | C15—N2—C11—C10 | −178.6 (3) |
| O1—Zn—N3—C6 | −79.5 (3) | Zn—N2—C11—C10 | 4.2 (4) |
| N2—Zn—N3—C6 | −176.8 (3) | N3—C10—C11—N2 | 1.4 (5) |
| N1—Zn—N3—C6 | −4.9 (2) | C9—C10—C11—N2 | 179.9 (3) |
| O4i—Zn—N3—C10 | −96.7 (2) | N3—C10—C11—C12 | −177.2 (3) |
| O1—Zn—N3—C10 | 104.2 (3) | C9—C10—C11—C12 | 1.3 (6) |
| N2—Zn—N3—C10 | 6.9 (2) | N2—C11—C12—C13 | −0.3 (6) |
| N1—Zn—N3—C10 | 178.8 (3) | C10—C11—C12—C13 | 178.2 (3) |
| C5—N1—C1—C2 | −0.3 (5) | C11—C12—C13—C14 | 0.1 (6) |
| Zn—N1—C1—C2 | −173.1 (3) | C12—C13—C14—C15 | 0.4 (6) |
| N1—C1—C2—C3 | −0.7 (6) | C11—N2—C15—C14 | 0.6 (6) |
| C1—C2—C3—C4 | 0.3 (6) | Zn—N2—C15—C14 | 177.3 (3) |
| C2—C3—C4—C5 | 0.9 (6) | C13—C14—C15—N2 | −0.7 (6) |
| Symmetry code: (i) −x+2, −y+2, −z+2. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1W—H1WB···O1ii | 0.81 (3) | 2.17 (3) | 2.957 (4) | 166 (4) |
| O1W—H1WA···O2i | 0.81 (3) | 2.04 (3) | 2.834 (4) | 169 (4) |
| Symmetry codes: (i) −x+2, −y+2, −z+2; (ii) x, y, z−1. |
Experimental details
| | (I) | (II) |
| Crystal data |
| Chemical formula | [Cd2(SO4)2(C16H12N6)2(H2O)2]·4H2O | [Cd2(SO4)2(C15H11N3)2]·2H2O |
| Mr | 1149.68 | 825.43 |
| Crystal system, space group | Triclinic, P1 | Triclinic, P1 |
| Temperature (K) | 293 | 293 |
| a, b, c (Å) | 8.870 (2), 10.640 (2), 12.685 (2) | 9.700 (2), 9.755 (2), 10.324 (2) |
| α, β, γ (°) | 103.36 (2), 98.79 (2), 110.45 (2) | 110.34 (2), 116.59 (2), 98.83 (2) |
| V (Å3) | 1055.0 (4) | 761.4 (4) |
| Z | 1 | 1 |
| Radiation type | Mo Kα | Mo Kα |
| µ (mm−1) | 1.19 | 1.79 |
| Crystal size (mm) | 0.20 × 0.12 × 0.05 | 0.24 × 0.16 × 0.10 |
| |
| Data collection |
| Diffractometer | Rigaku AFC-7S
diffractometer | Rigaku AFC-7S
diffractometer |
| Absorption correction | ψ scan
(MSC/AFC Diffractometer Control Software; Molecular Structure Corp, 1988) | ψ scan
(MSC/AFC Diffractometer Control Software; Molecular Structure Corp, 1988) |
| Tmin, Tmax | 0.83, 0.90 | 0.79, 0.86 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 5842, 4010, 1996 | 4216, 2890, 2008 |
| Rint | 0.039 | 0.039 |
| (sin θ/λ)max (Å−1) | 0.617 | 0.617 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.038, 0.056, 0.88 | 0.040, 0.073, 0.85 |
| No. of reflections | 4010 | 2890 |
| No. of parameters | 319 | 234 |
| No. of restraints | 13 | 3 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.44, −0.40 | 0.41, −0.35 |
Selected bond lengths (Å) for (I) top| Cd—O4i | 2.278 (3) | Cd—N3 | 2.437 (4) |
| Cd—N2 | 2.337 (4) | S—O3 | 1.465 (3) |
| Cd—O2 | 2.354 (3) | S—O2 | 1.463 (3) |
| Cd—O1W | 2.387 (4) | S—O1 | 1.464 (3) |
| Cd—O1 | 2.407 (3) | S—O4 | 1.465 (3) |
| Cd—N1 | 2.417 (4) | | |
| Symmetry code: (i) −x, −y, −z. |
Hydrogen-bond geometry (Å, º) for (I) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O3W—H3WA···O3i | 0.89 (2) | 1.90 (2) | 2.784 (5) | 170 (4) |
| O3W—H3WB···O3ii | 0.90 (2) | 1.91 (2) | 2.800 (4) | 171 (4) |
| O2W—H2WB···O4iii | 0.80 (2) | 2.34 (3) | 2.956 (5) | 134 (3) |
| O2W—H2WA···O3iii | 0.89 (2) | 2.50 (3) | 3.206 (5) | 137 (3) |
| O1W—H1WA···N6iv | 0.84 (2) | 2.21 (3) | 2.881 (5) | 136 (4) |
| O1W—H1WB···O3Wv | 0.86 (2) | 1.96 (3) | 2.791 (5) | 162 (4) |
| Symmetry codes: (i) −x, −y, −z; (ii) x, y+1, z; (iii) x+1, y+1, z+1; (iv) −x, −y, −z+1; (v) x, y−1, z. |
Selected bond lengths (Å) for (II) top| Zn—O4i | 1.952 (2) | S—O3 | 1.422 (3) |
| Zn—O1 | 1.970 (3) | S—O2 | 1.434 (3) |
| Zn—N3 | 2.074 (3) | S—O1 | 1.483 (3) |
| Zn—N2 | 2.166 (3) | S—O4 | 1.496 (2) |
| Zn—N1 | 2.201 (3) | | |
| Symmetry code: (i) −x+2, −y+2, −z+2. |
Hydrogen-bond geometry (Å, º) for (II) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1W—H1WB···O1ii | 0.81 (3) | 2.17 (3) | 2.957 (4) | 166 (4) |
| O1W—H1WA···O2i | 0.81 (3) | 2.04 (3) | 2.834 (4) | 169 (4) |
| Symmetry codes: (i) −x+2, −y+2, −z+2; (ii) x, y, z−1. |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu metal-organic compounds
metal-organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2003/05/00/ga1011/ga1011fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2003/05/00/ga1011/ga1011fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2003/05/00/ga1011/ga1011fig3thm.gif)

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry



The study of CdII and ZnII coordination compounds with sulfur oxoanions as ligands has been ongoing for many years. A number of structures, interesting from a stereochemical point of view, have been synthesized, as many of these anions (sulfite, sulfate, thiosulfate, tetrathionate, etc.) behave as versatile ligands when coordinating to d10 metal centers. The sulfate ion, in particular, is quite attractive in this respect; it has been reported as coordinating to transition metals through one, two or three O atoms, giving rise to monomeric, dimeric or polymeric structures and, in spite of extensive studies, the full binding capability of the anion does not seem to be exhausted.
A search in the November 2002 Version of the Cambridge Structural Database (CSD; Allen, 2002) returned 33 organometallic complexes in which the sulfate group is attached either to ZnII or CdII. The 16 ZnII complexes reported in the CSD are almost equally distributed between compounds displaying monomeric monodentate (M—O—SO3) and polymeric bridging (M—O—SO2—O—M) modes, with only three exceptions, which we will refer to by their CSD refcodes, viz. a dimer (MASVUX; Harvey et al., 2000), a one-dinemsional polymeric M—O—SO2—O—M structure (LOPNIN; Plater et al., 2000), where the sulfate group binds through three O atoms, two to the same metal center in a chelate fashion, and a polynuclear monomeric structure (VABMEK; Ali et al., 1998), in which the sulfate group binds to three different ZnII ions. Complexes with CdII, in contrast, show only a few examples of monomeric structures, the preferred coordination being polymeric of the M—O—SO2—O—M type. The only two exceptions [TIRXAT (Rahmani et al., 1996) and COXQUB (Huang et al., 1999)] organize as polymers built up by three O atoms, binding to three different metal centers. Continuing our research on this type of compounds, we report herein the structures of two new sulfate complexes, namely [Cd(SO4)(tpt)(H2O)·2H2O]2, (I), and [Zn(SO4)(tpy)·H2O]2, (II) [tpt is 2,4,6-tris(2-pyridyl)-1,3,5-triazine and tpy is 2,2':6',2''-terpyridine]. The former is the first report of a Cd(tpt) complex.
Both compounds crystallize in the space group P1 and form dimeric centrosymmetric structures. Complex (I) presents a heptacoordinated environment around the metal ion, formed by three N atoms from the tridentate tpt ligand, one aqua O atom and three O atoms from two symmetry-related sulfate ions, each of which binds to two metal centers, thus generating the dimeric structure (Fig. 1). In the two other Cd complexes incorporating tridentate sulfate ligands reported previously (TIRXAT and COXQUB), the anion binds to three different metal centers, generating polymeric structures. The coordination polyhedra in (I) can be described as a distorted pentagonal bipyramid, with the equatorial plane formed by atoms N1, N2, N3, O1 and O2 [maximum deviation of 0.04 (1) Å for N2], and angles subtended at the Cd site ranging from 58.6 (3) to 83.6 (3)° (ideal 72°). The axial sites are occupied by atoms O1W and O4(-x, −y, −z), and they subtend an angle of 166.1 (3)° to the cation.
The ligand is almost planar and acts in a tridentate manner, with the central Cd—N distance [2.337 (4) Å] being considerably shorter than the lateral Cd—N distances [2.437 (4) and 2.415 (4) Å]. Comparison with similar cases in the literature becomes difficult since there are no other reports of heptacoordinated cadmium(II) complexes with tpt or related tridentate ligands. There are, however, some examples where the heptacoordinated cation binds to a similar base [2,4-bis(4-pyridyl)-1,3,5-triazine] acting in a bridging manner, which display Cd—N distances in the range 2.28–2.32 Å (XESHOS, XESHUY and XESJAG; Barnett et al., 2001). The larger values in (I) could be attributed to the steric hindrance introduced by the planar tridentate bite.
Coordination does not affect the dimensions of the sulfate moiety; moreover, no significant differences are observed among the S—O distances involving either coordinated or non-coordinated O atoms. In fact, the mean S—O distance [1.463 (3) Å] is similar to that reported for the free anion [a search in the CSD gave a mean value of 1.472 (8) Å for 118 structures with R < 0.05]. The tpt ligand does not show any abnormal characteristics, with its three bound rings being basically coplanar; the atoms deviate from an ideal plane on average by less than 0.02 (1) Å. The terminal pyridyl ring deviates from planarity by less than 0.01 (1) Å and subtends an angle of 3.6 (1)° to the ideal plane. The structure is stabilized by hydrogen bonding with the two independent hydrate molecules (see Table 2).
The structure of (II) is also dimeric. The coordination is such that three N atoms from tpy and two O atoms from two symmetry-related sulfate ions complete a distorted fivefold environment around the metal ion (Fig. 2). The geometry lies midway between square pyramidal and trigonal bipyramidal. As in the case of (I), a considerable difference is observed in the Zn—N distances, that to the central N atom of tpy being the shortest [2.074 (3) versus 2.167 (3) and 2.201 (3) Å]. Similar results have been obtained in other pentacoordinated Zn–tpy complexes, viz. 2.209, 2.115 and 2.170 Å in DOLVIJ (Harrison et al., 1986), 2.181, 2.097 and 2.155 Å in OFABOM (Finn & Zubieta, 2002), and 2.169, 2.066 and 2.164 Å, and 2.158, 2.062 and 2.172 Å in XEZZIL (Hagrman & Zubieta, 2001).
As the zinc complex is pentacoordinated, larger valence bond values (Brown & Altermatt, 1985) for the Zn—O bonds are expected compared to those in the heptacoordinated cadmium complex, and, accordingly, a larger effect on the corresponding S—Ocoord distances of the sulfate group is predicted. In fact, the regular geometry displayed by the anion in (I) is lost in (II), where a clear difference is observed between the mean S—Ocoord and S—Ouncoord distances [1.490 (6) and 1.427 (6) Å, respectively]. Since (II) is the first reported structure containing pentacoordinated Zn cations bridged by sulfate anions in a Zn—O—SO2—O—Zn mode, it was not possible to make a fair comparison with other Zn complexes displaying analogous cation environments. We could trace, however, both tetra- and hexacoordinated Zn structures with a similar sulfate coordination. These structures clearly confirm the effect of bond valence; in the only tetracoordinated case of this sort reported so far (MASVUX; Harvey et al., 2000), the difference between the two types of S—O bonds is enhanced, being even larger than in the present pentacoordinated case [1.506 (6) and 1.439 (2) Å, respectively]; in the five hexacoordinated structures reported, in contrast, there is almost no trend differentiating the two kinds of bonds, their mean values being 1.47 (2) and 1.48 (3) Å, respectively, indistinguishable from each other and from the already mentioned average for the free ligand [1.472 (8) Å]. The geometry of the tpy ligand geometry and the internal parameters are unexceptional. There is one hydrogen-bonded crystallization water molecule in the structure (Table 4).
The packing is rather similar in both structures. As the dimers build up around a symmetry center, the planar molecules [tpt in (I) and tpy in (II)] are aligned parallel to each other. Cell symmetry preserves the orientation of the dimeric units, which stack with sets of parallel planes sharing one part of the double molecular units. This distribution (illustrated in Fig. 3) consists of a set of interleaved planar arrays at a nearly graphitic distance (3.4 Å). The linking agent is the extensive hydrogen-bonding network involving all the available water molecules acting as donors and, together with some N atoms of the organic ligand and sulfate O atoms, as acceptors, in some cases. The most important of these interactions are reported in Tables 2 and 4.