Buy article online - an online subscription or single-article purchase is required to access this article.
The title compound, C
8H
18N
4OP
+·I
− or [H
2cyclenPO][I], contains a five-coordinate P atom arranged in a slightly distorted trigonal bipyramid. The asymmetric unit contains half the cyclen moiety of the H~2~cyclenPO unit and two distinct iodide positions. The full molecule can be symmetry generated around the P atom using the symmetry code
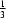
+
x −
y,
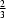
−
y,
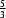
−
z. The P—N bond length in the equatorial plane is 1.656 (5) Å and the apical P—N interaction is much longer, 1.891 (4) Å. The cyclen moieties are linked together
via N—H

O hydrogen bonds and form a spiral chain along [001]. Crystallographically, there are two distinct anion sites, both in special positions, one of them with partial occupancy. One anion site may participate in an extremely weak hydrogen bond, linking the cyclen spiral chains into a pseudo-hexagonal three-dimensional array.
Supporting information
CCDC reference: 214645
Key indicators
- Single-crystal X-ray study
- T = 213 K
- Mean
![[sigma]](//journals.iucr.org/logos/entities/sigma_rmgif.gif) (C-C) = 0.010 Å
(C-C) = 0.010 Å
- Disorder in solvent or counterion
- R factor = 0.047
- wR factor = 0.116
- Data-to-parameter ratio = 18.1
checkCIF results
No syntax errors found
ADDSYM reports no extra symmetry
 Alert Level C:
Alert Level C:
PLAT_302 Alert C Anion/Solvent Disorder ....................... 34.00 Perc.
PLAT_601 Alert C Structure Contains Solvent Accessible VOIDS of 94.00 A 3
General Notes
REFLT_03
From the CIF: _diffrn_reflns_theta_max 25.36
From the CIF: _reflns_number_total 1324
Count of symmetry unique reflns 730
Completeness (_total/calc) 181.37%
TEST3: Check Friedels for noncentro structure
Estimate of Friedel pairs measured 594
Fraction of Friedel pairs measured 0.814
Are heavy atom types Z>Si present yes
Please check that the estimate of the number of Friedel pairs is
correct. If it is not, please give the correct count in the
_publ_section_exptl_refinement section of the submitted CIF.
0 Alert Level A = Potentially serious problem
0 Alert Level B = Potential problem
2 Alert Level C = Please check
Compound (I) was synthesized as a hydrolytic byproduct from reactions of cyclenphosphorane (Gupta et al., 1998). Colorless crystals were obtained from a CHCl3 solution, yield 95%. IR (cm−1, KBr pellet): 3100 (w), 2935 (versus), 2856 (s), 1460 (s), 1392 (versus), 1232 (s), 1141 (versus), 1088 (versus), 746 (m), 627 (s).
H atoms were positioned geometrically and refined using a riding model, with Uiso values constrained to be 1.2Ueq of the carrier atom. Atom H1 was located in a difference map. The distance was restrained and the Uiso value constrained to be 1.2Ueq of carrier atom N1. The compound was refined as a racemic twin.
Data collection: SMART (Bruker, 1997-1998); cell refinement: SAINT-Plus (Bruker, 1999); data reduction: SAINT-Plus; program(s) used to solve structure: XS in SHELXTL (Bruker, 1998); program(s) used to refine structure: XL in SHELXTL; molecular graphics: XP in SHELXTL; software used to prepare material for publication: XCIF in SHELXTL.
1,7-Dihydro-1,4,7,10-tetraazacyclododecanephosphine oxide iodide
top
Crystal data top
| C8H18IN4OP | Melting point: 230°C (dec.) K |
| Mr = 344.13 | Mo Kα radiation, λ = 0.71073 Å |
| Trigonal, R32 | Cell parameters from 3934 reflections |
| a = 19.327 (3) Å | θ = 2.3–23.5° |
| c = 10.052 (2) Å | µ = 2.31 mm−1 |
| V = 3251.7 (10) Å3 | T = 213 K |
| Z = 9 | Parallelepiped, colorless |
| F(000) = 1530 | 0.30 × 0.20 × 0.20 mm |
| Dx = 1.582 Mg m−3 | |
Data collection top
Burker/Siemens SMART 1K
diffractometer | 1324 independent reflections |
| Radiation source: normal-focus sealed tube | 1260 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.041 |
| Detector resolution: 8.3 pixels mm-1 | θmax = 25.4°, θmin = 2.4° |
| ω scans | h = −23→16 |
Absorption correction: multi-scan
(SADAB; Sheldrick, 1999) | k = −21→23 |
| Tmin = 0.522, Tmax = 0.628 | l = −12→11 |
| 14029 measured reflections | |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.047 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.117 | H-atom parameters constrained |
| S = 1.14 | w = 1/[σ2(Fo2) + (0.0627P)2 + 14.9452P]
where P = (Fo2 + 2Fc2)/3 |
| 1324 reflections | (Δ/σ)max < 0.001 |
| 73 parameters | Δρmax = 0.86 e Å−3 |
| 1 restraint | Δρmin = −0.86 e Å−3 |
Crystal data top
| C8H18IN4OP | Z = 9 |
| Mr = 344.13 | Mo Kα radiation |
| Trigonal, R32 | µ = 2.31 mm−1 |
| a = 19.327 (3) Å | T = 213 K |
| c = 10.052 (2) Å | 0.30 × 0.20 × 0.20 mm |
| V = 3251.7 (10) Å3 | |
Data collection top
Burker/Siemens SMART 1K
diffractometer | 1324 independent reflections |
Absorption correction: multi-scan
(SADAB; Sheldrick, 1999) | 1260 reflections with I > 2σ(I) |
| Tmin = 0.522, Tmax = 0.628 | Rint = 0.041 |
| 14029 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.047 | 1 restraint |
| wR(F2) = 0.117 | H-atom parameters constrained |
| S = 1.14 | w = 1/[σ2(Fo2) + (0.0627P)2 + 14.9452P]
where P = (Fo2 + 2Fc2)/3 |
| 1324 reflections | Δρmax = 0.86 e Å−3 |
| 73 parameters | Δρmin = −0.86 e Å−3 |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | Occ. (<1) |
| P1 | 0.90744 (10) | 0.3333 | 0.8333 | 0.0208 (5) | |
| O1 | 0.9861 (3) | 0.3333 | 0.8333 | 0.0205 (11) | |
| I1 | 0.6667 | 0.3333 | 0.8333 | 0.0380 (3) | |
| I1' | 0.49062 (10) | 0.3333 | 0.3333 | 0.0771 (5) | 0.67 |
| N1 | 0.8791 (3) | 0.2988 (3) | 1.0111 (4) | 0.0225 (10) | |
| H1 | 0.9201 | 0.3149 | 1.0620 | 0.027* | |
| N2 | 0.9082 (3) | 0.4160 (3) | 0.8791 (5) | 0.0259 (10) | |
| C2 | 0.8380 (4) | 0.3400 (4) | 1.0694 (6) | 0.0314 (13) | |
| H2A | 0.8412 | 0.3410 | 1.1667 | 0.038* | |
| H2B | 0.7816 | 0.3130 | 1.0430 | 0.038* | |
| C3 | 0.8830 (4) | 0.4243 (4) | 1.0126 (6) | 0.0348 (16) | |
| H3A | 0.8481 | 0.4476 | 1.0082 | 0.042* | |
| H3B | 0.9294 | 0.4587 | 1.0679 | 0.042* | |
| C4 | 0.9185 (4) | 0.4756 (4) | 0.7788 (6) | 0.0343 (15) | |
| H4A | 0.9555 | 0.5298 | 0.8106 | 0.041* | |
| H4B | 0.8672 | 0.4718 | 0.7576 | 0.041* | |
| C1 | 0.8298 (3) | 0.2109 (4) | 1.0083 (6) | 0.0317 (14) | |
| H1A | 0.7978 | 0.1912 | 1.0897 | 0.038* | |
| H1B | 0.8636 | 0.1866 | 1.0006 | 0.038* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| P1 | 0.0196 (7) | 0.0259 (11) | 0.0190 (9) | 0.0130 (5) | −0.0004 (4) | −0.0008 (7) |
| O1 | 0.0173 (19) | 0.024 (3) | 0.023 (3) | 0.0118 (13) | −0.0009 (10) | −0.002 (2) |
| I1 | 0.0285 (3) | 0.0285 (3) | 0.0570 (7) | 0.01423 (17) | 0.000 | 0.000 |
| I1' | 0.1195 (11) | 0.0398 (6) | 0.0453 (7) | 0.0199 (3) | 0.0013 (2) | 0.0026 (5) |
| N1 | 0.018 (2) | 0.029 (3) | 0.020 (2) | 0.012 (2) | −0.0017 (17) | 0.0021 (19) |
| N2 | 0.033 (3) | 0.028 (3) | 0.023 (2) | 0.020 (2) | 0.010 (2) | 0.004 (2) |
| C2 | 0.034 (3) | 0.043 (4) | 0.024 (3) | 0.024 (3) | 0.009 (3) | 0.001 (3) |
| C3 | 0.037 (3) | 0.045 (4) | 0.026 (3) | 0.023 (3) | 0.008 (3) | −0.002 (3) |
| C4 | 0.043 (4) | 0.037 (4) | 0.034 (3) | 0.029 (3) | 0.008 (3) | 0.009 (3) |
| C1 | 0.027 (3) | 0.036 (4) | 0.030 (3) | 0.013 (3) | 0.005 (2) | 0.010 (2) |
Geometric parameters (Å, º) top
| P1—O1 | 1.520 (5) | C2—H2A | 0.9800 |
| P1—N2i | 1.656 (5) | C2—H2B | 0.9800 |
| P1—N2 | 1.656 (5) | C3—H3A | 0.9800 |
| P1—N1i | 1.891 (4) | C3—H3B | 0.9800 |
| P1—N1 | 1.891 (4) | C4—C1i | 1.513 (8) |
| N1—C1 | 1.475 (8) | C4—H4A | 0.9800 |
| N1—C2 | 1.498 (7) | C4—H4B | 0.9800 |
| N1—H1 | 0.8595 | C1—C4i | 1.513 (8) |
| N2—C3 | 1.464 (7) | C1—H1A | 0.9800 |
| N2—C4 | 1.468 (7) | C1—H1B | 0.9800 |
| C2—C3 | 1.524 (9) | | |
| | | |
| O1—P1—N2i | 118.3 (2) | N1—C2—H2B | 110.8 |
| O1—P1—N2 | 118.3 (2) | C3—C2—H2B | 110.8 |
| N2i—P1—N2 | 123.4 (4) | H2A—C2—H2B | 108.9 |
| O1—P1—N1i | 96.47 (14) | N2—C3—C2 | 105.6 (5) |
| N2i—P1—N1i | 86.5 (2) | N2—C3—H3A | 110.6 |
| N2—P1—N1i | 87.3 (2) | C2—C3—H3A | 110.6 |
| O1—P1—N1 | 96.47 (14) | N2—C3—H3B | 110.6 |
| N2i—P1—N1 | 87.3 (2) | C2—C3—H3B | 110.6 |
| N2—P1—N1 | 86.5 (2) | H3A—C3—H3B | 108.7 |
| N1i—P1—N1 | 167.1 (3) | N2—C4—C1i | 104.9 (5) |
| C1—N1—C2 | 114.2 (4) | N2—C4—H4A | 110.8 |
| C1—N1—P1 | 107.2 (3) | C1i—C4—H4A | 110.8 |
| C2—N1—P1 | 108.6 (4) | N2—C4—H4B | 110.8 |
| C1—N1—H1 | 112.2 | C1i—C4—H4B | 110.8 |
| C2—N1—H1 | 102.3 | H4A—C4—H4B | 108.8 |
| P1—N1—H1 | 112.5 | N1—C1—C4i | 105.9 (5) |
| C3—N2—C4 | 117.7 (5) | N1—C1—H1A | 110.6 |
| C3—N2—P1 | 121.2 (4) | C4i—C1—H1A | 110.6 |
| C4—N2—P1 | 120.0 (4) | N1—C1—H1B | 110.6 |
| N1—C2—C3 | 104.6 (4) | C4i—C1—H1B | 110.6 |
| N1—C2—H2A | 110.8 | H1A—C1—H1B | 108.7 |
| C3—C2—H2A | 110.8 | | |
| Symmetry code: (i) x−y+1/3, −y+2/3, −z+5/3. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C2—H2B···I1 | 0.98 | 3.23 | 4.023 (6) | 140 |
| N1—H1···O1ii | 0.86 | 1.91 | 2.769 (5) | 176 |
| Symmetry code: (ii) −x+y+5/3, −x+4/3, z+1/3. |
Experimental details
| Crystal data |
| Chemical formula | C8H18IN4OP |
| Mr | 344.13 |
| Crystal system, space group | Trigonal, R32 |
| Temperature (K) | 213 |
| a, c (Å) | 19.327 (3), 10.052 (2) |
| V (Å3) | 3251.7 (10) |
| Z | 9 |
| Radiation type | Mo Kα |
| µ (mm−1) | 2.31 |
| Crystal size (mm) | 0.30 × 0.20 × 0.20 |
| |
| Data collection |
| Diffractometer | Burker/Siemens SMART 1K
diffractometer |
| Absorption correction | Multi-scan
(SADAB; Sheldrick, 1999) |
| Tmin, Tmax | 0.522, 0.628 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 14029, 1324, 1260 |
| Rint | 0.041 |
| (sin θ/λ)max (Å−1) | 0.603 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.047, 0.117, 1.14 |
| No. of reflections | 1324 |
| No. of parameters | 73 |
| No. of restraints | 1 |
| H-atom treatment | H-atom parameters constrained |
| w = 1/[σ2(Fo2) + (0.0627P)2 + 14.9452P]
where P = (Fo2 + 2Fc2)/3 |
| Δρmax, Δρmin (e Å−3) | 0.86, −0.86 |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C2—H2B···I1 | 0.98 | 3.23 | 4.023 (6) | 140 |
| N1—H1···O1i | 0.86 | 1.91 | 2.769 (5) | 176 |
| Symmetry code: (i) −x+y+5/3, −x+4/3, z+1/3. |

 Subscribe to Acta Crystallographica Section E: Crystallographic Communications
Subscribe to Acta Crystallographica Section E: Crystallographic Communications
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu organic compounds
organic compounds














![[Figure 1]](http://journals.iucr.org/e/issues/2003/05/00/fl6031/fl6031fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/e/issues/2003/05/00/fl6031/fl6031fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/e/issues/2003/05/00/fl6031/fl6031fig3thm.gif)




Cyclen chemistry is extensive with 431 hits in the November 2002 issue of the Cambridge Structural Database (CSD; Allen, 2002). However, only one cyclen phosphine oxide is described in the database, namely cyclenphosphine oxide dihydrate, (II) (Oget et al., 1999). Compound (II) was first described in a patent (Richman, 1976) and the solution structure elucidated by NMR methods (Richman & Kubale, 1983). Although other cyclen phosphine chalcogenide derivatives have been synthesized (Oget et al., 1998), no structural information has been reported.
Compound (I) is a hydrolytic by-product formed during reactions of cyclenphosphorane (Gupta et al., 1998). The title compound, (I), is similar to (II). (I) is protonated, cationic and charge balanced by an iodide anion (see Fig. 1). The P atom lies on a special position and the other half of the molecule is symmetry generated. The P atom is five-coordinate and forms a slightly distorted trigonal bipyramid (tbp). The equatorial plane [N2, O1, N2i and P1; symmtery code: (i) 1/3 + x-y, 2/3 − y, 5/3 − z] is planar, with the sum of the angles around P1 equal to 360°. The distortion from tbp can be seen in the axial and equatorial N—P—N angles [167.1 (3), 123.4 (4) and 118.3 (2)°]. The P—N distances show that the equatorial N atoms are bonded to the phosphorus [P1—N2 and P1—N2i = 1.656 (5) Å]. The apical P—N distances [P1—N1 and P1—Nii = 1.891 (4) Å] are longer than a single P—N bond. They are considerably shorter than the sum of the van der Waals radii (3.35 Å) and indicate a strong coordination by these N atoms to the central phosphorus. The elongation of the apical P—N bond in a tbp is expected and this bond lengthening has been reported previously with a range of P—Napical distances of 1.8 to 2.11 Å [e.g. CSD refcode YAFDUE (Khasnis et al., 1992), LOBYUW (Gupta et al., 1999) and GOCPUJ (Oget at al., 1999)]. Atoms N1 and N1i are both protonated and the H atom was located on the difference map. The I atom is site shared in the asymmetric unit in two distinct special positions, with occupancies of 1/6 for I1 and 1/3 for I1'. I1 is located at Wyckoff position b (multiplicity 3) at full occupancy. I1' is located at position d (multiplicity 9) at 0.6667 of full occupancy for charge balance, and in keeping with the refined occupancy (0.16666 for I1 and 0.3333 for I1').
The cyclen phosphine oxide moiety is twisted along the N1—P1—N1i plane. The amido H atoms are oriented to either side of this plane (0.41 and −0.41 Å). This produces a slight helical distortion of the cyclen moiety. This helical distortion can also be seen in bis(borane)cyclenphosphorane (Dupart et al., 1985). The absolute configuration of chiral (I) could not be determined reliably and (I) was refined as a racemic mixture.
The extended solid state structure generated by intermolecular interactions presents an interesting picture:
(i) N—H···O hydrogen bonding. There are two of these interactions per cyclen moiety. They link the cyclen PO units into a spiral chain that runs parallel to [001]. This is shown in Figs. 2(a) and 2(b). Distances and angles are given in Table 1.
(ii) C—H···I interactions. These `interactions' should be considered extremely weak or almost non-existant. They are at the furthest limit of reported weak C—H···X interactions (Desiraju & Steiner, 2001). However, they may have an influence on the supramolecular association by linking the spiral cyclen chains into a pseudo-hexagonal array (three H2cyclenPO cations and three I− anions) which is shown in Fig. 3. The space between these networked molecules are occupied by the symmetry related unassociated iodine position. This leaves large voids between the spiral chains, ca 94 Å3 in the asymmetric unit.