Details of the structures of two conformational polymorphs of the title compound, C
12H
17N
2OS
+·Cl
−, are reported. In form (I) (space group
P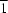
), the two N—H groups of the cation are in a
trans conformation, while in form (II) (space group
P2
1/
c), they are in a
cis arrangement. This results in different packing and hydrogen-bond arrangements in the two forms, both of which have extended chains lying along the
a direction. In form (I), these chains are composed of centrosymmetric
R42(18) (N—H

Cl and O—H
Cl) hydrogen-bonded rings and
R22(18) (N—H
O) hydrogen-bonded rings. In form (II), the chains are formed by centrosymmetric
R42(18) (N—H
Cl and O—H
Cl) hydrogen-bonded rings and by
R42(12) (N—H
Cl) hydrogen-bonded rings.
Supporting information
CCDC references: 810016; 810017
3,5-Dimethyl-4-isothiocyanatophenol (1.70 g, 9.50 mmol) was dissolved in dry
dichloromethane (20 ml) and 3-aminopropanol (1.70 ml, 22.18 mmol) was added.
The reaction mixture was refluxed overnight with stirring. The solution was
then cooled down to room temperature and the solvent removed under reduced
pressure to give the crude product. Concentrated hydrochloric acid solution (8 ml) was added to the crude product and the resulting solution was refluxed
overnight with stirring. The solution was poured into 10% NaOH (50 ml) and
stirred for 3 h. The final product (2.0 g, yield 90.9%) was precipitated using
Dowex resin H+ form (pH = 1) (Kai et al., 2007). Crystals from
methanol and ethanol were found to be the same and were designated as form
(I), and those from propan-2-ol were form (II).
All H atoms were found in difference Fourier maps and were subsequently placed
in idealized positions, with O—H = 0.82 Å, N—H = 0.86 Å,
Csp2—H = 0.93 Å, Csp3—H = 0.97 Å for CH2 H atoms
and 0.96 Å for methyl H atoms. All H atoms were allowed for as riding, with
Uiso(H) = 1.5Ueq(parent atom) for O—H and methyl H atoms,
and 1.2Ueq(parent atom) for all others.
For both compounds, data collection: COLLECT (Nonius, 2002); cell refinement: DENZO-SMN (Otwinowski & Minor, 1997); data reduction: DENZO-SMN (Otwinowski & Minor, 1997); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008); molecular graphics: XP in SHELXTL (Sheldrick, 2008); software used to prepare material for publication: SHELXL97 (Sheldrick, 2008) and local procedures.
(I) 2-(4-hydroxy-2,6-dimethylanilino)-5,6-dihydro-4
H-1,3-thiazin-3-ium
chloride
top
Crystal data top
| C12H17N2OS+·Cl− | Z = 2 |
| Mr = 272.79 | F(000) = 288 |
| Triclinic, P1 | Dx = 1.376 Mg m−3 |
| a = 6.9961 (1) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 7.9421 (1) Å | Cell parameters from 2964 reflections |
| c = 13.0864 (2) Å | θ = 1.0–27.5° |
| α = 73.3925 (6)° | µ = 0.44 mm−1 |
| β = 84.1579 (6)° | T = 90 K |
| γ = 70.8388 (6)° | Block, colourless |
| V = 658.17 (2) Å3 | 0.30 × 0.20 × 0.10 mm |
Data collection top
Nonius KappaCCD area-detector
diffractometer | 2981 independent reflections |
| Radiation source: fine-focus sealed tube | 2762 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.017 |
| Detector resolution: 18 pixels mm-1 | θmax = 27.5°, θmin = 1.6° |
| ω scans at fixed χ = 55° | h = −9→9 |
Absorption correction: multi-scan
(SCALEPACK; Otwinowski & Minor, 1997) | k = −10→10 |
| Tmin = 0.881, Tmax = 0.958 | l = −16→16 |
| 5923 measured reflections | |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.028 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.072 | H-atom parameters constrained |
| S = 1.03 | w = 1/[σ2(Fo2) + (0.028P)2 + 0.3783P]
where P = (Fo2 + 2Fc2)/3 |
| 2981 reflections | (Δ/σ)max = 0.001 |
| 156 parameters | Δρmax = 0.32 e Å−3 |
| 0 restraints | Δρmin = −0.28 e Å−3 |
Crystal data top
| C12H17N2OS+·Cl− | γ = 70.8388 (6)° |
| Mr = 272.79 | V = 658.17 (2) Å3 |
| Triclinic, P1 | Z = 2 |
| a = 6.9961 (1) Å | Mo Kα radiation |
| b = 7.9421 (1) Å | µ = 0.44 mm−1 |
| c = 13.0864 (2) Å | T = 90 K |
| α = 73.3925 (6)° | 0.30 × 0.20 × 0.10 mm |
| β = 84.1579 (6)° | |
Data collection top
Nonius KappaCCD area-detector
diffractometer | 2981 independent reflections |
Absorption correction: multi-scan
(SCALEPACK; Otwinowski & Minor, 1997) | 2762 reflections with I > 2σ(I) |
| Tmin = 0.881, Tmax = 0.958 | Rint = 0.017 |
| 5923 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.028 | 0 restraints |
| wR(F2) = 0.072 | H-atom parameters constrained |
| S = 1.03 | Δρmax = 0.32 e Å−3 |
| 2981 reflections | Δρmin = −0.28 e Å−3 |
| 156 parameters | |
Special details top
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell esds are taken
into account individually in the estimation of esds in distances, angles
and torsion angles; correlations between esds in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell esds is used for estimating esds involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and
goodness of fit S are based on F2, conventional R-factors R are based
on F, with F set to zero for negative F2. The threshold expression of
F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is
not relevant to the choice of reflections for refinement. R-factors based
on F2 are statistically about twice as large as those based on F, and R-
factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| S1 | 0.33096 (5) | −0.11563 (4) | 0.90302 (2) | 0.01881 (9) | |
| O4 | 0.24327 (13) | 0.72281 (12) | 0.36708 (7) | 0.01848 (19) | |
| H4 | 0.2369 | 0.6870 | 0.3134 | 0.028* | |
| N2 | 0.23495 (15) | 0.17820 (14) | 0.74414 (8) | 0.0162 (2) | |
| H2 | 0.1171 | 0.1916 | 0.7773 | 0.019* | |
| N3 | 0.57340 (15) | 0.01012 (14) | 0.74502 (8) | 0.0158 (2) | |
| H3 | 0.5868 | 0.0909 | 0.6849 | 0.019* | |
| C2 | 0.39103 (18) | 0.03369 (16) | 0.78804 (9) | 0.0143 (2) | |
| C4 | 0.75611 (19) | −0.14085 (17) | 0.78973 (10) | 0.0198 (3) | |
| H4A | 0.8745 | −0.1024 | 0.7646 | 0.024* | |
| H4B | 0.7671 | −0.2478 | 0.7651 | 0.024* | |
| C5 | 0.7503 (2) | −0.1936 (2) | 0.91032 (11) | 0.0259 (3) | |
| H5A | 0.7327 | −0.0853 | 0.9351 | 0.031* | |
| H5B | 0.8779 | −0.2849 | 0.9375 | 0.031* | |
| C6 | 0.5788 (2) | −0.2721 (2) | 0.95266 (11) | 0.0293 (3) | |
| H6A | 0.5769 | −0.3014 | 1.0299 | 0.035* | |
| H6B | 0.6043 | −0.3865 | 0.9331 | 0.035* | |
| C11 | 0.24705 (17) | 0.31409 (16) | 0.64568 (9) | 0.0139 (2) | |
| C12 | 0.24680 (17) | 0.27297 (16) | 0.54866 (10) | 0.0141 (2) | |
| C13 | 0.24924 (17) | 0.40896 (16) | 0.45421 (9) | 0.0147 (2) | |
| H13 | 0.2524 | 0.3833 | 0.3889 | 0.018* | |
| C14 | 0.24696 (17) | 0.58294 (16) | 0.45753 (9) | 0.0148 (2) | |
| C15 | 0.24651 (17) | 0.62147 (16) | 0.55438 (10) | 0.0158 (2) | |
| H15 | 0.2451 | 0.7385 | 0.5553 | 0.019* | |
| C16 | 0.24809 (17) | 0.48781 (16) | 0.65008 (10) | 0.0148 (2) | |
| C17 | 0.23777 (19) | 0.08723 (16) | 0.54606 (10) | 0.0176 (2) | |
| H17A | 0.2302 | 0.0861 | 0.4734 | 0.026* | |
| H17B | 0.1203 | 0.0652 | 0.5847 | 0.026* | |
| H17C | 0.3571 | −0.0079 | 0.5783 | 0.026* | |
| C18 | 0.2548 (2) | 0.52859 (18) | 0.75508 (10) | 0.0201 (3) | |
| H18A | 0.3847 | 0.4595 | 0.7872 | 0.030* | |
| H18B | 0.1510 | 0.4938 | 0.8017 | 0.030* | |
| H18C | 0.2331 | 0.6585 | 0.7432 | 0.030* | |
| Cl1 | −0.18978 (4) | 0.32705 (4) | 0.84515 (2) | 0.02133 (9) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| S1 | 0.02351 (17) | 0.01550 (15) | 0.01458 (15) | −0.00706 (12) | 0.00309 (11) | 0.00028 (11) |
| O4 | 0.0215 (4) | 0.0158 (4) | 0.0162 (4) | −0.0084 (3) | 0.0005 (3) | 0.0015 (3) |
| N2 | 0.0154 (5) | 0.0150 (5) | 0.0146 (5) | −0.0034 (4) | 0.0035 (4) | −0.0012 (4) |
| N3 | 0.0164 (5) | 0.0125 (5) | 0.0146 (5) | −0.0031 (4) | 0.0008 (4) | 0.0003 (4) |
| C2 | 0.0190 (6) | 0.0121 (5) | 0.0127 (5) | −0.0056 (4) | 0.0008 (4) | −0.0040 (4) |
| C4 | 0.0166 (6) | 0.0165 (6) | 0.0202 (6) | −0.0009 (5) | 0.0007 (5) | −0.0011 (5) |
| C5 | 0.0250 (7) | 0.0256 (7) | 0.0191 (6) | 0.0020 (5) | −0.0049 (5) | −0.0040 (5) |
| C6 | 0.0304 (7) | 0.0217 (7) | 0.0187 (6) | 0.0035 (6) | 0.0048 (5) | 0.0054 (5) |
| C11 | 0.0118 (5) | 0.0124 (5) | 0.0144 (5) | −0.0026 (4) | 0.0007 (4) | −0.0004 (4) |
| C12 | 0.0111 (5) | 0.0123 (5) | 0.0175 (6) | −0.0032 (4) | 0.0016 (4) | −0.0031 (4) |
| C13 | 0.0130 (5) | 0.0162 (6) | 0.0143 (5) | −0.0042 (4) | 0.0013 (4) | −0.0043 (4) |
| C14 | 0.0106 (5) | 0.0141 (5) | 0.0167 (6) | −0.0044 (4) | 0.0007 (4) | 0.0007 (4) |
| C15 | 0.0134 (5) | 0.0117 (5) | 0.0222 (6) | −0.0041 (4) | −0.0016 (4) | −0.0037 (5) |
| C16 | 0.0110 (5) | 0.0155 (5) | 0.0172 (6) | −0.0023 (4) | −0.0008 (4) | −0.0050 (4) |
| C17 | 0.0199 (6) | 0.0140 (6) | 0.0194 (6) | −0.0064 (5) | 0.0024 (5) | −0.0049 (5) |
| C18 | 0.0206 (6) | 0.0204 (6) | 0.0195 (6) | −0.0042 (5) | −0.0022 (5) | −0.0077 (5) |
| Cl1 | 0.01833 (16) | 0.02361 (16) | 0.01703 (15) | −0.00342 (12) | 0.00454 (11) | −0.00310 (12) |
Geometric parameters (Å, º) top
| S1—C2 | 1.7394 (12) | C6—H6B | 0.9700 |
| S1—C6 | 1.8237 (15) | C11—C12 | 1.3988 (17) |
| O4—C14 | 1.3680 (14) | C11—C16 | 1.3997 (16) |
| O4—H4 | 0.8395 | C12—C13 | 1.3925 (16) |
| N2—C2 | 1.3296 (16) | C12—C17 | 1.5071 (16) |
| N2—C11 | 1.4405 (14) | C13—C14 | 1.3897 (16) |
| N2—H2 | 0.8803 | C13—H13 | 0.9300 |
| N3—C2 | 1.3180 (16) | C14—C15 | 1.3857 (17) |
| N3—C4 | 1.4693 (15) | C15—C16 | 1.3895 (17) |
| N3—H3 | 0.8802 | C15—H15 | 0.9300 |
| C4—C5 | 1.5125 (18) | C16—C18 | 1.5076 (17) |
| C4—H4A | 0.9700 | C17—H17A | 0.9600 |
| C4—H4B | 0.9700 | C17—H17B | 0.9600 |
| C5—C6 | 1.515 (2) | C17—H17C | 0.9600 |
| C5—H5A | 0.9700 | C18—H18A | 0.9600 |
| C5—H5B | 0.9700 | C18—H18B | 0.9600 |
| C6—H6A | 0.9700 | C18—H18C | 0.9600 |
| | | |
| C2—S1—C6 | 102.79 (6) | C12—C11—N2 | 119.48 (10) |
| C14—O4—H4 | 109.5 | C16—C11—N2 | 118.61 (10) |
| C2—N2—C11 | 123.96 (10) | C13—C12—C11 | 118.70 (11) |
| C2—N2—H2 | 118.0 | C13—C12—C17 | 120.48 (11) |
| C11—N2—H2 | 118.0 | C11—C12—C17 | 120.80 (10) |
| C2—N3—C4 | 124.96 (10) | C14—C13—C12 | 120.01 (11) |
| C2—N3—H3 | 117.5 | C14—C13—H13 | 120.0 |
| C4—N3—H3 | 117.5 | C12—C13—H13 | 120.0 |
| N3—C2—N2 | 121.02 (11) | O4—C14—C15 | 117.25 (10) |
| N3—C2—S1 | 124.62 (9) | O4—C14—C13 | 122.26 (11) |
| N2—C2—S1 | 114.37 (9) | C15—C14—C13 | 120.49 (11) |
| N3—C4—C5 | 111.27 (10) | C14—C15—C16 | 120.96 (11) |
| N3—C4—H4A | 109.4 | C14—C15—H15 | 119.5 |
| C5—C4—H4A | 109.4 | C16—C15—H15 | 119.5 |
| N3—C4—H4B | 109.4 | C15—C16—C11 | 117.99 (11) |
| C5—C4—H4B | 109.4 | C15—C16—C18 | 120.68 (11) |
| H4A—C4—H4B | 108.0 | C11—C16—C18 | 121.32 (11) |
| C4—C5—C6 | 110.83 (12) | C12—C17—H17A | 109.5 |
| C4—C5—H5A | 109.5 | C12—C17—H17B | 109.5 |
| C6—C5—H5A | 109.5 | H17A—C17—H17B | 109.5 |
| C4—C5—H5B | 109.5 | C12—C17—H17C | 109.5 |
| C6—C5—H5B | 109.5 | H17A—C17—H17C | 109.5 |
| H5A—C5—H5B | 108.1 | H17B—C17—H17C | 109.5 |
| C5—C6—S1 | 113.60 (9) | C16—C18—H18A | 109.5 |
| C5—C6—H6A | 108.8 | C16—C18—H18B | 109.5 |
| S1—C6—H6A | 108.8 | H18A—C18—H18B | 109.5 |
| C5—C6—H6B | 108.8 | C16—C18—H18C | 109.5 |
| S1—C6—H6B | 108.8 | H18A—C18—H18C | 109.5 |
| H6A—C6—H6B | 107.7 | H18B—C18—H18C | 109.5 |
| C12—C11—C16 | 121.82 (11) | | |
| | | |
| C4—N3—C2—N2 | 177.58 (11) | C16—C11—C12—C17 | −177.69 (11) |
| C4—N3—C2—S1 | −2.90 (17) | N2—C11—C12—C17 | −1.09 (16) |
| C11—N2—C2—N3 | 3.03 (18) | C11—C12—C13—C14 | −1.54 (17) |
| C11—N2—C2—S1 | −176.54 (9) | C17—C12—C13—C14 | 176.66 (11) |
| C6—S1—C2—N3 | 9.14 (12) | C12—C13—C14—O4 | −178.06 (10) |
| C6—S1—C2—N2 | −171.30 (10) | C12—C13—C14—C15 | 1.35 (17) |
| C2—N3—C4—C5 | −34.24 (17) | O4—C14—C15—C16 | 179.37 (10) |
| N3—C4—C5—C6 | 64.57 (14) | C13—C14—C15—C16 | −0.07 (18) |
| C4—C5—C6—S1 | −57.07 (14) | C14—C15—C16—C11 | −0.95 (17) |
| C2—S1—C6—C5 | 20.83 (12) | C14—C15—C16—C18 | 177.89 (11) |
| C2—N2—C11—C12 | 78.84 (15) | C12—C11—C16—C15 | 0.74 (17) |
| C2—N2—C11—C16 | −104.46 (13) | N2—C11—C16—C15 | −175.89 (10) |
| C16—C11—C12—C13 | 0.50 (17) | C12—C11—C16—C18 | −178.09 (11) |
| N2—C11—C12—C13 | 177.10 (10) | N2—C11—C16—C18 | 5.28 (17) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H2···Cl1 | 0.88 | 2.28 | 3.1244 (11) | 161 |
| N3—H3···O4i | 0.88 | 2.12 | 2.8143 (13) | 136 |
| O4—H4···Cl1ii | 0.84 | 2.17 | 2.9913 (10) | 165 |
| Symmetry codes: (i) −x+1, −y+1, −z+1; (ii) −x, −y+1, −z+1. |
(II) 2-(4-hydroxy-2,6-dimethylanilino)-5,6-dihydro-4
H-1,3-thiazin-3-ium
chloride
top
Crystal data top
| C12H17N2OS+·Cl− | F(000) = 576 |
| Mr = 272.79 | Dx = 1.323 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 11.8877 (2) Å | Cell parameters from 3332 reflections |
| b = 9.2120 (2) Å | θ = 1.0–27.5° |
| c = 12.6673 (3) Å | µ = 0.42 mm−1 |
| β = 99.0242 (10)° | T = 90 K |
| V = 1370.02 (5) Å3 | Rod, colourless |
| Z = 4 | 0.50 × 0.20 × 0.10 mm |
Data collection top
Nonius KappaCCD area-detector
diffractometer | 3141 independent reflections |
| Radiation source: fine-focus sealed tube | 2389 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.031 |
| Detector resolution: 18 pixels mm-1 | θmax = 27.5°, θmin = 1.7° |
| ω scans at fixed χ = 55° | h = −15→15 |
Absorption correction: multi-scan
(SCALEPACK; Otwinowski & Minor, 1997) | k = −11→11 |
| Tmin = 0.818, Tmax = 0.959 | l = −16→16 |
| 6046 measured reflections | |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.042 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.112 | H-atom parameters constrained |
| S = 1.10 | w = 1/[σ2(Fo2) + (0.0534P)2 + 0.5868P]
where P = (Fo2 + 2Fc2)/3 |
| 3141 reflections | (Δ/σ)max = 0.001 |
| 157 parameters | Δρmax = 0.50 e Å−3 |
| 0 restraints | Δρmin = −0.31 e Å−3 |
Crystal data top
| C12H17N2OS+·Cl− | V = 1370.02 (5) Å3 |
| Mr = 272.79 | Z = 4 |
| Monoclinic, P21/c | Mo Kα radiation |
| a = 11.8877 (2) Å | µ = 0.42 mm−1 |
| b = 9.2120 (2) Å | T = 90 K |
| c = 12.6673 (3) Å | 0.50 × 0.20 × 0.10 mm |
| β = 99.0242 (10)° | |
Data collection top
Nonius KappaCCD area-detector
diffractometer | 3141 independent reflections |
Absorption correction: multi-scan
(SCALEPACK; Otwinowski & Minor, 1997) | 2389 reflections with I > 2σ(I) |
| Tmin = 0.818, Tmax = 0.959 | Rint = 0.031 |
| 6046 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.042 | 0 restraints |
| wR(F2) = 0.112 | H-atom parameters constrained |
| S = 1.10 | Δρmax = 0.50 e Å−3 |
| 3141 reflections | Δρmin = −0.31 e Å−3 |
| 157 parameters | |
Special details top
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell esds are taken
into account individually in the estimation of esds in distances, angles
and torsion angles; correlations between esds in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell esds is used for estimating esds involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and
goodness of fit S are based on F2, conventional R-factors R are based
on F, with F set to zero for negative F2. The threshold expression of
F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is
not relevant to the choice of reflections for refinement. R-factors based
on F2 are statistically about twice as large as those based on F, and R-
factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| S1 | 0.26830 (4) | 0.87420 (5) | 0.70402 (4) | 0.01887 (15) | |
| O4 | −0.19758 (11) | 0.58513 (17) | 0.64205 (11) | 0.0213 (3) | |
| H4 | −0.2399 | 0.6219 | 0.5919 | 0.032* | |
| N2 | 0.25741 (14) | 0.63610 (17) | 0.59182 (13) | 0.0177 (4) | |
| H2 | 0.2845 | 0.5673 | 0.5574 | 0.021* | |
| N3 | 0.43335 (13) | 0.74466 (18) | 0.61184 (13) | 0.0166 (4) | |
| H3 | 0.4541 | 0.6768 | 0.5724 | 0.020* | |
| C2 | 0.32677 (17) | 0.7434 (2) | 0.62978 (15) | 0.0160 (4) | |
| C4 | 0.51844 (17) | 0.8530 (2) | 0.65420 (17) | 0.0177 (4) | |
| H4A | 0.5797 | 0.8531 | 0.6118 | 0.021* | |
| H4B | 0.5505 | 0.8280 | 0.7271 | 0.021* | |
| C5 | 0.46578 (17) | 1.0029 (2) | 0.65208 (17) | 0.0194 (4) | |
| H5A | 0.4280 | 1.0246 | 0.5803 | 0.023* | |
| H5B | 0.5252 | 1.0746 | 0.6717 | 0.023* | |
| C6 | 0.38039 (17) | 1.0115 (2) | 0.72899 (17) | 0.0213 (5) | |
| H6A | 0.3456 | 1.1070 | 0.7236 | 0.026* | |
| H6B | 0.4203 | 1.0001 | 0.8014 | 0.026* | |
| C11 | 0.13924 (16) | 0.6285 (2) | 0.60511 (16) | 0.0153 (4) | |
| C12 | 0.05724 (17) | 0.7029 (2) | 0.53393 (15) | 0.0170 (4) | |
| C13 | −0.05634 (17) | 0.6893 (2) | 0.54664 (15) | 0.0172 (4) | |
| H13 | −0.1123 | 0.7388 | 0.5009 | 0.021* | |
| C14 | −0.08699 (16) | 0.6022 (2) | 0.62725 (15) | 0.0162 (4) | |
| C15 | −0.00477 (17) | 0.5284 (2) | 0.69634 (15) | 0.0172 (4) | |
| H15 | −0.0263 | 0.4704 | 0.7498 | 0.021* | |
| C16 | 0.10955 (17) | 0.5402 (2) | 0.68661 (15) | 0.0164 (4) | |
| C17 | 0.09010 (19) | 0.7947 (3) | 0.44518 (17) | 0.0247 (5) | |
| H17A | 0.0230 | 0.8204 | 0.3963 | 0.037* | |
| H17B | 0.1408 | 0.7408 | 0.4079 | 0.037* | |
| H17C | 0.1274 | 0.8813 | 0.4747 | 0.037* | |
| C18 | 0.19913 (17) | 0.4591 (2) | 0.76046 (17) | 0.0219 (5) | |
| H18A | 0.1637 | 0.4040 | 0.8106 | 0.033* | |
| H18B | 0.2521 | 0.5268 | 0.7984 | 0.033* | |
| H18C | 0.2389 | 0.3945 | 0.7196 | 0.033* | |
| Cl1 | 0.38915 (4) | 0.35624 (5) | 0.54403 (4) | 0.01989 (15) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| S1 | 0.0162 (3) | 0.0169 (3) | 0.0252 (3) | −0.00198 (19) | 0.0083 (2) | −0.0064 (2) |
| O4 | 0.0135 (7) | 0.0278 (8) | 0.0228 (8) | −0.0018 (6) | 0.0031 (6) | 0.0059 (6) |
| N2 | 0.0170 (9) | 0.0164 (9) | 0.0213 (9) | −0.0009 (7) | 0.0076 (7) | −0.0061 (7) |
| N3 | 0.0146 (9) | 0.0152 (8) | 0.0213 (9) | −0.0007 (7) | 0.0066 (7) | −0.0045 (7) |
| C2 | 0.0178 (10) | 0.0152 (10) | 0.0155 (10) | 0.0001 (8) | 0.0045 (8) | 0.0009 (8) |
| C4 | 0.0144 (10) | 0.0185 (10) | 0.0206 (10) | −0.0008 (8) | 0.0044 (8) | −0.0017 (8) |
| C5 | 0.0190 (10) | 0.0147 (10) | 0.0259 (11) | −0.0024 (8) | 0.0074 (9) | −0.0002 (8) |
| C6 | 0.0180 (10) | 0.0163 (10) | 0.0302 (12) | −0.0034 (8) | 0.0058 (9) | −0.0069 (9) |
| C11 | 0.0147 (10) | 0.0138 (10) | 0.0182 (10) | −0.0021 (7) | 0.0052 (8) | −0.0040 (8) |
| C12 | 0.0215 (10) | 0.0141 (10) | 0.0157 (10) | −0.0039 (8) | 0.0043 (8) | −0.0034 (8) |
| C13 | 0.0181 (10) | 0.0172 (10) | 0.0159 (10) | 0.0017 (8) | 0.0009 (8) | 0.0000 (8) |
| C14 | 0.0149 (10) | 0.0188 (10) | 0.0151 (9) | −0.0028 (8) | 0.0030 (8) | −0.0043 (8) |
| C15 | 0.0200 (10) | 0.0169 (10) | 0.0154 (9) | −0.0034 (8) | 0.0046 (8) | −0.0016 (8) |
| C16 | 0.0179 (10) | 0.0139 (10) | 0.0174 (10) | −0.0014 (8) | 0.0024 (8) | −0.0042 (8) |
| C17 | 0.0257 (12) | 0.0243 (12) | 0.0246 (11) | −0.0042 (9) | 0.0052 (9) | 0.0042 (9) |
| C18 | 0.0192 (11) | 0.0243 (12) | 0.0222 (11) | 0.0017 (9) | 0.0034 (8) | 0.0023 (9) |
| Cl1 | 0.0199 (3) | 0.0203 (3) | 0.0197 (3) | 0.00485 (19) | 0.0041 (2) | −0.00182 (19) |
Geometric parameters (Å, º) top
| S1—C2 | 1.739 (2) | C6—H6B | 0.9700 |
| S1—C6 | 1.828 (2) | C11—C12 | 1.399 (3) |
| O4—C14 | 1.366 (2) | C11—C16 | 1.403 (3) |
| O4—H4 | 0.8200 | C12—C13 | 1.390 (3) |
| N2—C2 | 1.328 (2) | C12—C17 | 1.506 (3) |
| N2—C11 | 1.443 (2) | C13—C14 | 1.392 (3) |
| N2—H2 | 0.8600 | C13—H13 | 0.9300 |
| N3—C2 | 1.322 (2) | C14—C15 | 1.384 (3) |
| N3—C4 | 1.462 (2) | C15—C16 | 1.388 (3) |
| N3—H3 | 0.8600 | C15—H15 | 0.9300 |
| C4—C5 | 1.514 (3) | C16—C18 | 1.502 (3) |
| C4—H4A | 0.9700 | C17—H17A | 0.9600 |
| C4—H4B | 0.9700 | C17—H17B | 0.9600 |
| C5—C6 | 1.515 (3) | C17—H17C | 0.9600 |
| C5—H5A | 0.9700 | C18—H18A | 0.9600 |
| C5—H5B | 0.9700 | C18—H18B | 0.9600 |
| C6—H6A | 0.9700 | C18—H18C | 0.9600 |
| | | |
| C2—S1—C6 | 103.33 (10) | C12—C11—N2 | 119.61 (17) |
| C14—O4—H4 | 109.5 | C16—C11—N2 | 118.43 (17) |
| C2—N2—C11 | 123.66 (16) | C13—C12—C11 | 118.20 (18) |
| C2—N2—H2 | 118.2 | C13—C12—C17 | 120.48 (18) |
| C11—N2—H2 | 118.2 | C11—C12—C17 | 121.32 (18) |
| C2—N3—C4 | 124.70 (16) | C12—C13—C14 | 120.51 (18) |
| C2—N3—H3 | 117.6 | C12—C13—H13 | 119.7 |
| C4—N3—H3 | 117.6 | C14—C13—H13 | 119.7 |
| N3—C2—N2 | 120.09 (17) | O4—C14—C15 | 117.15 (17) |
| N3—C2—S1 | 124.21 (15) | O4—C14—C13 | 122.38 (18) |
| N2—C2—S1 | 115.69 (15) | C15—C14—C13 | 120.47 (18) |
| N3—C4—C5 | 110.84 (16) | C14—C15—C16 | 120.66 (18) |
| N3—C4—H4A | 109.5 | C14—C15—H15 | 119.7 |
| C5—C4—H4A | 109.5 | C16—C15—H15 | 119.7 |
| N3—C4—H4B | 109.5 | C15—C16—C11 | 118.26 (18) |
| C5—C4—H4B | 109.5 | C15—C16—C18 | 120.96 (18) |
| H4A—C4—H4B | 108.1 | C11—C16—C18 | 120.77 (18) |
| C4—C5—C6 | 110.68 (17) | C12—C17—H17A | 109.5 |
| C4—C5—H5A | 109.5 | C12—C17—H17B | 109.5 |
| C6—C5—H5A | 109.5 | H17A—C17—H17B | 109.5 |
| C4—C5—H5B | 109.5 | C12—C17—H17C | 109.5 |
| C6—C5—H5B | 109.5 | H17A—C17—H17C | 109.5 |
| H5A—C5—H5B | 108.1 | H17B—C17—H17C | 109.5 |
| C5—C6—S1 | 113.33 (14) | C16—C18—H18A | 109.5 |
| C5—C6—H6A | 108.9 | C16—C18—H18B | 109.5 |
| S1—C6—H6A | 108.9 | H18A—C18—H18B | 109.5 |
| C5—C6—H6B | 108.9 | C16—C18—H18C | 109.5 |
| S1—C6—H6B | 108.9 | H18A—C18—H18C | 109.5 |
| H6A—C6—H6B | 107.7 | H18B—C18—H18C | 109.5 |
| C12—C11—C16 | 121.89 (18) | | |
| | | |
| C4—N3—C2—N2 | −176.75 (17) | C16—C11—C12—C17 | −178.56 (18) |
| C4—N3—C2—S1 | 2.0 (3) | N2—C11—C12—C17 | −1.8 (3) |
| C11—N2—C2—N3 | −178.18 (17) | C11—C12—C13—C14 | −0.9 (3) |
| C11—N2—C2—S1 | 3.0 (2) | C17—C12—C13—C14 | 178.68 (18) |
| C6—S1—C2—N3 | 7.4 (2) | C12—C13—C14—O4 | −179.57 (18) |
| C6—S1—C2—N2 | −173.80 (15) | C12—C13—C14—C15 | 0.4 (3) |
| C2—N3—C4—C5 | −39.1 (3) | O4—C14—C15—C16 | −179.95 (17) |
| N3—C4—C5—C6 | 66.5 (2) | C13—C14—C15—C16 | 0.1 (3) |
| C4—C5—C6—S1 | −56.1 (2) | C14—C15—C16—C11 | 0.0 (3) |
| C2—S1—C6—C5 | 19.88 (18) | C14—C15—C16—C18 | −179.26 (18) |
| C2—N2—C11—C12 | 84.1 (2) | C12—C11—C16—C15 | −0.6 (3) |
| C2—N2—C11—C16 | −99.1 (2) | N2—C11—C16—C15 | −177.36 (16) |
| C16—C11—C12—C13 | 1.0 (3) | C12—C11—C16—C18 | 178.68 (18) |
| N2—C11—C12—C13 | 177.78 (17) | N2—C11—C16—C18 | 1.9 (3) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H2···Cl1 | 0.86 | 2.33 | 3.1245 (17) | 154 |
| N3—H3···Cl1i | 0.86 | 2.57 | 3.2437 (17) | 136 |
| O4—H4···Cl1ii | 0.82 | 2.28 | 3.0579 (14) | 159 |
| Symmetry codes: (i) −x+1, −y+1, −z+1; (ii) −x, −y+1, −z+1. |
Experimental details
| | (I) | (II) |
| Crystal data |
| Chemical formula | C12H17N2OS+·Cl− | C12H17N2OS+·Cl− |
| Mr | 272.79 | 272.79 |
| Crystal system, space group | Triclinic, P1 | Monoclinic, P21/c |
| Temperature (K) | 90 | 90 |
| a, b, c (Å) | 6.9961 (1), 7.9421 (1), 13.0864 (2) | 11.8877 (2), 9.2120 (2), 12.6673 (3) |
| α, β, γ (°) | 73.3925 (6), 84.1579 (6), 70.8388 (6) | 90, 99.0242 (10), 90 |
| V (Å3) | 658.17 (2) | 1370.02 (5) |
| Z | 2 | 4 |
| Radiation type | Mo Kα | Mo Kα |
| µ (mm−1) | 0.44 | 0.42 |
| Crystal size (mm) | 0.30 × 0.20 × 0.10 | 0.50 × 0.20 × 0.10 |
| |
| Data collection |
| Diffractometer | Nonius KappaCCD area-detector
diffractometer | Nonius KappaCCD area-detector
diffractometer |
| Absorption correction | Multi-scan
(SCALEPACK; Otwinowski & Minor, 1997) | Multi-scan
(SCALEPACK; Otwinowski & Minor, 1997) |
| Tmin, Tmax | 0.881, 0.958 | 0.818, 0.959 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 5923, 2981, 2762 | 6046, 3141, 2389 |
| Rint | 0.017 | 0.031 |
| (sin θ/λ)max (Å−1) | 0.649 | 0.649 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.028, 0.072, 1.03 | 0.042, 0.112, 1.10 |
| No. of reflections | 2981 | 3141 |
| No. of parameters | 156 | 157 |
| H-atom treatment | H-atom parameters constrained | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.32, −0.28 | 0.50, −0.31 |
Hydrogen-bond geometry (Å, º) for (I) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H2···Cl1 | 0.88 | 2.28 | 3.1244 (11) | 161 |
| N3—H3···O4i | 0.88 | 2.12 | 2.8143 (13) | 136 |
| O4—H4···Cl1ii | 0.84 | 2.17 | 2.9913 (10) | 165 |
| Symmetry codes: (i) −x+1, −y+1, −z+1; (ii) −x, −y+1, −z+1. |
Hydrogen-bond geometry (Å, º) for (II) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H2···Cl1 | 0.86 | 2.33 | 3.1245 (17) | 154 |
| N3—H3···Cl1i | 0.86 | 2.57 | 3.2437 (17) | 136 |
| O4—H4···Cl1ii | 0.82 | 2.28 | 3.0579 (14) | 159 |
| Symmetry codes: (i) −x+1, −y+1, −z+1; (ii) −x, −y+1, −z+1. |


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2010/12/00/fg3203/fg3203fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2010/12/00/fg3203/fg3203fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2010/12/00/fg3203/fg3203fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/c/issues/2010/12/00/fg3203/fg3203fig4thm.gif)




Polymorphism, the phenomenon of a given molecule existing in more than one crystal structure, is a normal observation for organics (McCrone, 1965). Polymorphism is of great importance in pharmaceuticals as well as in materials science, because individual forms may have different physicochemical properties which can potentially lead to new formulations or new materials. Conformational polymorphism, a branch of polymorphism, is particularly interesting since it provides ideal cases for structure–property relationship studies (Bernstein, 2002, 1987). Conformational polymorphism arises from intrinsic molecular flexibility and is the result of a compromise between inter- and intramolecular interactions.
The free base of the title compound, (1), is the principal metabolite fragment recovered from equine urine after enzymatic hydrolysis of xylazine [N-(2,6-dimethylphenyl)-5,6-dihydro-4H-1,3-thiazin-2-amine], which is a relatively short-acting α-2 agonist tranquilizer widely used in equine medicine. Optimal regulatory control of the use of xylazine is dependent on the detection and quantification of urinary metabolites or metabolite fragments such as the free base of (1) (Mutlib et al., 1992). We report here the conformational polymorphism of (1), which occurs in two crystalline forms, viz. (I) and (II).
Our analysis establishes that form (I) is triclinic (space group P1) and form (II) monoclinic (space group P21/c), with one formula unit in the asymmetric unit in each case. Views of forms (I) and (II) are given in Figs. 1 and 2, respectively. In both forms, imine atom N3 is protonated, and in both forms the six-membered heterocyclic ring has a half-chair conformation, with atom C5 0.704 (2) Å from the S1/C2/N3/C4/C6 plane in form (I) and 0.699 (2) Å from the same plane in form (II). The C2—N2 and C2—N3 bond lengths in (I) are 1.3296 (16) and 1.3180 (16) Å, respectively, and the corresponding values in (II) are 1.328 (2) and 1.322 (2) Å. These dimensions are entirely consistent with delocalization of the C2═C3 double bond over the C2—N2—C3 moiety, as shown in structures (1a) and (1b) in the scheme. Thus, the two cations could either be considered as configurational isomers (with C2—N2 considered as a double bond), with form (I) the E isomer and form (II) the Z isomer, or as conformational isomers (with C2—N3 considered as the double bond). As seen in Figs. 1 and 2 (which have been drawn to have similar orientations of the six-membered heterocyclic rings), the principal difference between the two forms is in the orientation of the 4-hydroxy-2,6-dimethylaniline moiety with respect to the heterocyclic ring. In form (I), the S1—C2—N2—C11 torsion angle is -176.54 (9)°, while in form (II) the corresponding value is 3.0 (2)°.
Due to the conformational difference between the cations in the two polymorphs, the packing patterns are dissimilar. In polymorph (I) (Fig. 3), hydrogen bonds between the protonated imine NH group and the phenol O atom (N3—H3···O4i; see Table 1 for details) link two cations to form an 18-membered ring dimer centred at (1/2, 1/2, 1/2), with the hydrogen-bonded ring graph-set descriptor R22(18) (Bernstein et al., 1995). This dimer is further connected through hydrogen bonds between the Cl- ion and the hydroxy O4—H4 and secondary N2–H2 groups (details in Table 1) to form a second set of 18-membered rings, but this time with hydrogen-bond descriptor R42(18), lying about inversion centres at (0, 1/2, 1/2), (1, 1/2, 1/2), etc. This gives rise to a one-dimensional chain along the a axis of the triclinic cell.
In form (II) (Fig. 4), the cations are interconnected through hydrogen bonds between the Cl- ion and all three hydrogen-bonding donors, viz. O4—H4, imine N3—H3 and amino N2—H2 (details in Table 2). There is a 12-membered ring [centred at (1/2, 1/2, 1/2)] involving the N2—H2 and N3—H3 groups and the Cl- ion, with descriptor R42(12) (details in Table 2). This dimer is then connected via N2—H2···Cl1 and O4—H4···Cl1ii hydrogen bonds (Table 2) to generate 18-membered rings [centred at (0, 1/2, 1/2), (1, 1/2, 1/2), etc.] with descriptor R42(18), the same as in form (I). In this way, a one-dimensional chain is developed along the a axis of this monoclinic cell.
Our work has thus shown that the two crystalline forms discovered for (1) can either be considered as configurational or conformational isomers, due to the delocalisation of the amine lone-pair electrons over three atoms. The configurational/conformational variation in the two forms gives rise to differences in packing and hydrogen-bond arrangements in the crystal structures.