Buy article online - an online subscription or single-article purchase is required to access this article.
An ice-like hexameric water cluster, stabilized by the flexible bis-imidazolyl compound 2,3,5,6-tetrafluoro-1,4-bis(imidazol-1-ylmethyl)benzene (Fbix), is found in the trigonal
R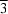
crystal structure of the title compound, C
14H
10F
4N
4·2H
2O or Fbix·2H
2O. The Fbix molecule lies about an inversion centre with one water molecule in the asymmetric unit in a general position. A cyclic chair-like hexameric water cluster with
symmetry is generated with a hydrogen-bonded O

O distance within the hexamer of 2.786 (3) Å. The Fbix molecule adopts a
trans conformation, where the imidazole ring makes a dihedral angle of 70.24 (11)° with the central tetrafluorinated aromatic ring. Each water hexamer is connected by six Fbix molecules through intermolecular O—H
N hydrogen bonds [N
O = 2.868 (3) Å] to yield a three-dimensional supramolecular network with primitive cubic (pcu) topology. Large voids in each single pcu network lead to fourfold interpenetrated aggregates of Fbix·2H
2O.
Supporting information
CCDC reference: 774888
The compound Fbix was synthesized according to the literature method described
by Gao et al. (2006), through the reaction of
1,4-bis-(chloromethyl)-2,3,5,6-tetrafluorobenzene with extensive imidazole in
methanol solution (yield circa 60% on the basis of the former).
Colourless block single crystals of the hydrate, (I), were obtained by
recrystallizing the products from aqueous solution at room temperature.
All H atoms bound to C atoms were assigned to calculated positions, with C—H
= 0.93 (aromatic) and 0.97 Å (methylene) and refined using a riding model,
with Uiso(H) = 1.2Ueq(C). The H atoms of water were firstly
located in a difference Fourier map and then refined as riding with the
restraints of O—H = 0.82 Å and H···H = 1.43 Å [Uiso(H) =
1.5Ueq(O)].
Data collection: APEX2 (Bruker, 2007); cell refinement: APEX2 and SAINT (Bruker, 2007); data reduction: SAINT (Bruker, 2007); program(s) used to solve structure: SHELXTL (Sheldrick, 2008); program(s) used to refine structure: SHELXTL (Sheldrick, 2008); molecular graphics: SHELXTL (Sheldrick, 2008) and DIAMOND (Brandenburg, 2005); software used to prepare material for publication: SHELXTL (Sheldrick, 2008).
Crystal data top
| C14H10F4N4·2H2O | Dx = 1.486 Mg m−3 |
| Mr = 346.29 | Mo Kα radiation, λ = 0.71073 Å |
| Trigonal, R3 | Cell parameters from 5189 reflections |
| Hall symbol: -R 3 | θ = 3.1–32.2° |
| a = 17.753 (10) Å | µ = 0.13 mm−1 |
| c = 12.756 (7) Å | T = 297 K |
| V = 3482 (3) Å3 | Block, colorless |
| Z = 9 | 0.24 × 0.22 × 0.22 mm |
| F(000) = 1602 | |
Data collection top
Bruker APEXII CCD
diffractometer | 1518 independent reflections |
| Radiation source: fine-focus sealed tube | 1327 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.018 |
| ϕ and ω scans | θmax = 25.9°, θmin = 2.1° |
Absorption correction: multi-scan
(SADABS; Sheldrick, 2003) | h = −17→21 |
| Tmin = 0.969, Tmax = 0.971 | k = −18→21 |
| 8266 measured reflections | l = −15→6 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.042 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.134 | H-atom parameters constrained |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.0865P)2 + 1.7362P]
where P = (Fo2 + 2Fc2)/3 |
| 1518 reflections | (Δ/σ)max < 0.001 |
| 109 parameters | Δρmax = 0.40 e Å−3 |
| 0 restraints | Δρmin = −0.22 e Å−3 |
Crystal data top
| C14H10F4N4·2H2O | Z = 9 |
| Mr = 346.29 | Mo Kα radiation |
| Trigonal, R3 | µ = 0.13 mm−1 |
| a = 17.753 (10) Å | T = 297 K |
| c = 12.756 (7) Å | 0.24 × 0.22 × 0.22 mm |
| V = 3482 (3) Å3 | |
Data collection top
Bruker APEXII CCD
diffractometer | 1518 independent reflections |
Absorption correction: multi-scan
(SADABS; Sheldrick, 2003) | 1327 reflections with I > 2σ(I) |
| Tmin = 0.969, Tmax = 0.971 | Rint = 0.018 |
| 8266 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.042 | 0 restraints |
| wR(F2) = 0.134 | H-atom parameters constrained |
| S = 1.05 | Δρmax = 0.40 e Å−3 |
| 1518 reflections | Δρmin = −0.22 e Å−3 |
| 109 parameters | |
Special details top
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are
estimated using the full covariance matrix. The cell esds are taken into
account individually in the estimation of esds in distances, angles and
torsion angles; correlations between esds in cell parameters are only used
when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell esds is used for estimating esds involving l.s. planes. |
Refinement. Refinement of F^2^ against ALL reflections. The weighted
R-factor wR and goodness of fit S are based on F^2^,
conventional R-factors R are based on F, with F
set to zero for negative F^2^. The threshold expression of
F^2^ > σ(F^2^) is used only for calculating
R-factors(gt) etc. and is not relevant to the choice of reflections for
refinement. R-factors based on F^2^ are statistically about
twice as large as those based on F, and R- factors based on ALL
data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| O1 | 0.16893 (10) | 0.07977 (9) | 0.03938 (10) | 0.0743 (4) | |
| H1A | 0.1886 | 0.0955 | 0.0986 | 0.111* | |
| H1B | 0.1396 | 0.0978 | 0.0109 | 0.111* | |
| C1 | 0.18731 (11) | 0.16251 (11) | 0.42122 (15) | 0.0578 (4) | |
| H1 | 0.1638 | 0.1663 | 0.4848 | 0.069* | |
| C2 | 0.14391 (14) | 0.12350 (12) | 0.33282 (18) | 0.0715 (6) | |
| H2 | 0.0837 | 0.0952 | 0.3256 | 0.086* | |
| C3 | 0.27711 (14) | 0.17490 (13) | 0.29756 (13) | 0.0639 (5) | |
| H3 | 0.3287 | 0.1902 | 0.2624 | 0.077* | |
| C4 | 0.34753 (10) | 0.24816 (10) | 0.46431 (11) | 0.0502 (4) | |
| H4A | 0.3522 | 0.3044 | 0.4751 | 0.060* | |
| H4B | 0.4001 | 0.2576 | 0.4294 | 0.060* | |
| C5 | 0.34011 (9) | 0.20595 (9) | 0.56866 (10) | 0.0434 (4) | |
| C6 | 0.37128 (9) | 0.14992 (10) | 0.58466 (11) | 0.0454 (4) | |
| C7 | 0.30183 (10) | 0.22127 (10) | 0.65303 (11) | 0.0458 (4) | |
| F1 | 0.40904 (7) | 0.13179 (7) | 0.50588 (8) | 0.0648 (3) | |
| F2 | 0.27087 (7) | 0.27585 (7) | 0.64270 (8) | 0.0638 (3) | |
| N1 | 0.27302 (8) | 0.19521 (8) | 0.39768 (10) | 0.0480 (4) | |
| N2 | 0.20007 (14) | 0.13113 (12) | 0.25527 (14) | 0.0776 (5) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| O1 | 0.0931 (10) | 0.0857 (10) | 0.0558 (8) | 0.0535 (8) | −0.0088 (6) | −0.0084 (6) |
| C1 | 0.0526 (9) | 0.0532 (9) | 0.0623 (10) | 0.0224 (7) | −0.0069 (7) | 0.0043 (7) |
| C2 | 0.0675 (12) | 0.0525 (9) | 0.0875 (14) | 0.0247 (9) | −0.0329 (10) | −0.0023 (9) |
| C3 | 0.0868 (13) | 0.0758 (11) | 0.0440 (9) | 0.0518 (11) | −0.0165 (8) | −0.0104 (7) |
| C4 | 0.0486 (8) | 0.0545 (9) | 0.0397 (8) | 0.0201 (7) | −0.0055 (6) | 0.0028 (6) |
| C5 | 0.0389 (7) | 0.0493 (8) | 0.0361 (7) | 0.0177 (6) | −0.0060 (5) | −0.0017 (5) |
| C6 | 0.0426 (7) | 0.0555 (8) | 0.0360 (7) | 0.0230 (6) | −0.0025 (5) | −0.0060 (6) |
| C7 | 0.0444 (8) | 0.0494 (8) | 0.0452 (8) | 0.0247 (6) | −0.0065 (6) | −0.0034 (6) |
| F1 | 0.0764 (7) | 0.0877 (7) | 0.0445 (6) | 0.0517 (6) | 0.0086 (4) | −0.0024 (4) |
| F2 | 0.0775 (7) | 0.0752 (7) | 0.0594 (6) | 0.0536 (6) | −0.0013 (5) | 0.0022 (5) |
| N1 | 0.0547 (8) | 0.0517 (7) | 0.0398 (7) | 0.0282 (6) | −0.0101 (5) | −0.0017 (5) |
| N2 | 0.1045 (14) | 0.0774 (11) | 0.0632 (10) | 0.0547 (10) | −0.0401 (10) | −0.0225 (8) |
Geometric parameters (Å, º) top
| O1—H1A | 0.8202 | C4—N1 | 1.4533 (19) |
| O1—H1B | 0.8203 | C4—C5 | 1.501 (2) |
| C1—C2 | 1.346 (3) | C4—H4A | 0.9700 |
| C1—N1 | 1.364 (2) | C4—H4B | 0.9700 |
| C1—H1 | 0.9300 | C5—C7 | 1.371 (2) |
| C2—N2 | 1.362 (3) | C5—C6 | 1.374 (2) |
| C2—H2 | 0.9300 | C6—F1 | 1.3336 (17) |
| C3—N2 | 1.305 (3) | C6—C7i | 1.370 (2) |
| C3—N1 | 1.339 (2) | C7—F2 | 1.3383 (18) |
| C3—H3 | 0.9300 | C7—C6i | 1.370 (2) |
| | | |
| H1A—O1—H1B | 121.3 | H4A—C4—H4B | 107.9 |
| C2—C1—N1 | 105.33 (18) | C7—C5—C6 | 116.49 (13) |
| C2—C1—H1 | 127.3 | C7—C5—C4 | 121.72 (14) |
| N1—C1—H1 | 127.3 | C6—C5—C4 | 121.79 (13) |
| C1—C2—N2 | 110.79 (18) | F1—C6—C7i | 118.78 (14) |
| C1—C2—H2 | 124.6 | F1—C6—C5 | 119.72 (13) |
| N2—C2—H2 | 124.6 | C7i—C6—C5 | 121.50 (14) |
| N2—C3—N1 | 111.90 (19) | F2—C7—C6i | 118.63 (14) |
| N2—C3—H3 | 124.1 | F2—C7—C5 | 119.36 (13) |
| N1—C3—H3 | 124.1 | C6i—C7—C5 | 122.01 (14) |
| N1—C4—C5 | 111.81 (12) | C3—N1—C1 | 107.14 (14) |
| N1—C4—H4A | 109.3 | C3—N1—C4 | 125.28 (15) |
| C5—C4—H4A | 109.3 | C1—N1—C4 | 127.42 (14) |
| N1—C4—H4B | 109.3 | C3—N2—C2 | 104.84 (16) |
| C5—C4—H4B | 109.3 | | |
| | | |
| N1—C1—C2—N2 | −0.1 (2) | C4—C5—C7—C6i | −179.55 (13) |
| N1—C4—C5—C7 | −90.79 (17) | N2—C3—N1—C1 | −0.3 (2) |
| N1—C4—C5—C6 | 89.18 (17) | N2—C3—N1—C4 | −175.97 (15) |
| C7—C5—C6—F1 | 179.76 (13) | C2—C1—N1—C3 | 0.22 (19) |
| C4—C5—C6—F1 | −0.2 (2) | C2—C1—N1—C4 | 175.78 (14) |
| C7—C5—C6—C7i | −0.5 (2) | C5—C4—N1—C3 | −131.29 (16) |
| C4—C5—C6—C7i | 179.55 (13) | C5—C4—N1—C1 | 53.9 (2) |
| C6—C5—C7—F2 | 179.42 (13) | N1—C3—N2—C2 | 0.2 (2) |
| C4—C5—C7—F2 | −0.6 (2) | C1—C2—N2—C3 | −0.1 (2) |
| C6—C5—C7—C6i | 0.5 (2) | | |
| Symmetry code: (i) −x+2/3, −y+1/3, −z+4/3. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1A···N2 | 0.82 | 2.08 | 2.867 (3) | 162 |
| O1—H1B···O1ii | 0.82 | 1.98 | 2.786 (3) | 166 |
| C4—H4B···O1iii | 0.97 | 2.47 | 3.404 (3) | 161 |
| Symmetry codes: (ii) x−y, x, −z; (iii) −x+2/3, −y+1/3, −z+1/3. |
Experimental details
| Crystal data |
| Chemical formula | C14H10F4N4·2H2O |
| Mr | 346.29 |
| Crystal system, space group | Trigonal, R3 |
| Temperature (K) | 297 |
| a, c (Å) | 17.753 (10), 12.756 (7) |
| V (Å3) | 3482 (3) |
| Z | 9 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.13 |
| Crystal size (mm) | 0.24 × 0.22 × 0.22 |
| |
| Data collection |
| Diffractometer | Bruker APEXII CCD
diffractometer |
| Absorption correction | Multi-scan
(SADABS; Sheldrick, 2003) |
| Tmin, Tmax | 0.969, 0.971 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 8266, 1518, 1327 |
| Rint | 0.018 |
| (sin θ/λ)max (Å−1) | 0.615 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.042, 0.134, 1.05 |
| No. of reflections | 1518 |
| No. of parameters | 109 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.40, −0.22 |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1A···N2 | 0.82 | 2.08 | 2.867 (3) | 162 |
| O1—H1B···O1i | 0.82 | 1.98 | 2.786 (3) | 166 |
| C4—H4B···O1ii | 0.97 | 2.47 | 3.404 (3) | 161 |
| Symmetry codes: (i) x−y, x, −z; (ii) −x+2/3, −y+1/3, −z+1/3. |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2010/04/00/fg3149/fg3149fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2010/04/00/fg3149/fg3149fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2010/04/00/fg3149/fg3149fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/c/issues/2010/04/00/fg3149/fg3149fig4thm.gif)

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry



In recent years there have been many systematic and successful studies of the crystal engineering of organic crystals, and evidence has been published demonstrating the molecule recognitions of hydrogen-bonding and/or other noncolvalent interactions (Desiraju, 1995; Hosseini, 2005). In the field of water chemistry, intense efforts have focused on unravelling the structural morphologies of water aggregates to improve understanding of the nature of water–water interactions in bulk water or ice (Ludwig, 2001). Small water clusters, (H2O)n where n = 3–8, have been extensively studied and structurally characterized; numerous water clusters including tetramers (Zhang, Tian et al., 2007; Xu et al., 2008), pentamers (Ma et al., 2004), hexamers (Ye et al., 2004; Mukhopadhyay & Bernal, 2005; Siddiqui et al., 2008) and octamers (Doedens et al., 2002) reveal various conformations in the crystal hosts in the solid state. Among these, the water hexamer is of great interest since it represents the smallest possible unit that can exhibit some of the properties found in bulk water (Gregory et al., 1997). Moreover, the water hexamer behaves as the transition from two-dimensional to three-dimensional structures by adopting ring (chair and boat), book, bag, cage, and prism with nearly equal energy by the energetic discrimination of 0.7 kcal.mol-1 (Ludwig, 2000, 2001). This realization has prompted considerable attention on the structural characterization of water hexamers trapped in hydrate clathrates (Moorthy et al., 2002; Mukhopadhyay & Bernal, 2005).
Imidazole-containing ligands such as 1,4-bis(imidazol-1-yl-methyl)benzene (bix) (Hoskins et al., 1997a,b; Fan et al., 2005), 1,1-(1,4-butanediyl)bis(imidazole) (bbix) (Ma et al., 2000; Duncan et al., 1996), 1,3,5-tris(imidazole-1-yl-methyl)benzene (tib) (Liu & Tong, 2002) and related species have been used to generate a rich variety of metal–organic architectures with interesting structural topologies and potential properties, as well as many hydrogen-bonding aggregates (Aakeröy et al., 2005, 2006; Zhang, Gembicky et al., 2007; Xu et al., 2008). Considering the weak intermolecular forces among fluorinated compounds, Shreeve and co-workers have reported three novel coordination polymers constructed from transition metals with 2,3,5,6-tetrafluoro-1,4-bis(imidazol-1-yl-methyl)benzene (Fbix) (Gao et al., 2006). In the process of exploring the structural diversity and recognition pattern of the Fbix building block, we isolated its dihydrate, Fbix.2H2O, (I). Herein, we describe the formation and structural features of the cyclic water hexamer with an ice-like chair conformation and its assembly into a cubic supramolecular network through the O—H···N linkages with the Fbix crystal host.
The asymmetric unit of (I) comprises one Fbix on an inversion centre at (1/3,1/6,2/3) and one lattice water in a general position near the unit-cell origin; a perspective view with the atom-numbering scheme is shown in Fig. 1. The Fbix molecule adopts a trans conformation with a dihedral angle of 70.24 (11)° between the terminal imidazole ring and the central tetrafluorinated benzene plane. The R-3 space-group symmetry generates a hexameric cluster of water with -3 symmetry and a perfect chair form of the O atoms (Fig. 2). Within the chair-like hexameric cluster, each water molecule is simultaneously involved in interactions with symmetry-related water molecules (see Table 1) leading to a head-to-tail R66(12) hydrogen-bonding pattern (Etter, 1990). The hydrogen-bonded O···O distance within the water hexamer [2.786 (3) Å], is comparable with the value in ice Ih (2.759 Å) at 183 K (Eisenberg & Kauzmann, 1969) and shorter than the value in liquid water (2.85 Å) (Narten et al., 1982). The O—O—O—O torsion angles in the hexamer are ±63.54 (2)° and the O—O—O angle [107.95 (7)°] is close to the corresponding value of 109.3° for the preferred tetrahedral geometry in hexagonal ice. By way of contrast, a similar water hexamer crystallizing in the same R-3 space group as (I) has an almost ideal planar system (Moorthy, et al., 2002) with an O···O distance of 2.906 (5) Å, an O—O—O bond angle of 120.0 (2)° and O—O—O—O torsion angles of ±3.0 (2)°.
In the present system, the other H atom of each water molecule is connected to the Fbix moiety via an O1—H1A···N2 hydrogen bond (see Table 1). From the viewpoint of topology, each discrete cyclic water hexamer, acting as a six-connected node, is linked by the bi-connected Fbix spacer to generate a three-dimensional supramolecular network (Fig. 3). The topology is as follows: the motif of the water hexamer connected to six Fbix linkers is represented to join other six hexamers through the O—H···N interactions, which results in a six-connected net with the Schläfli symbol 412.63 (that represents primitive cubic topology), the net being somewhat offset. Because of the large void in each single net with the adjoining hexameric centre-to-centre distance separated by the Fbix linker [19.858 (11) Å], the final fourfold interpenetrated structure of this type is realized through a translation vector [1/3,-1/3,-1/3] of circa 11.10 Å according to the calculation in TOPOS (Blatov, 2004), as illustrated in Fig. 4. All the independent nets are related by this single vector and the whole fourfold interpenetrated array is generated by translating the primitive single net three times, which has a PIC (primitive interpenetration cell) with the unit-cell vectors [4/3,-4/3,-4/3], [0,1,0] and [1,0,0] (Blatov, 2004).