Buy article online - an online subscription or single-article purchase is required to access this article.
The title compound, C
6H
12O
4, also known as dimeric acetone peroxide, Me
2(C
2O
4)Me
2, has crystallographically imposed inversion symmetry and adopts a chair conformation in the solid state. This structure contrasts with that of the sulfur homologue Me
2(C
2S
4)Me
2, which has crystallographically imposed
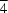
symmetry and crystallizes in a twist-boat conformation. Crystals of the title compound are twinned along the reciprocal
c* axis.
Supporting information
CCDC reference: 235337
Compound (I) is an explosive solid and the recommended safety precautions should be taken into account before attempting to prepare it (Dankowski & Prescher, 1988). Since Baeyer & Villiger (1899, 1900) prepared (I) by the treatment of acetone with Caro's acid (peroxomonosulfuric acid, H2SO5), many other methods have been developed to synthesize this molecule, including ozonolysis of tetramethylethylene (Criegee et al., 1953; Milas et al., 1955; Lockley et al., 2000), treatment of acetone with concentrated H2O2 in acetonitrile acidified with H2SO4 (McCullough et al., 1980) or reaction of acetone with bis(trimethylsilyl)peroxide (Jefford & Boukouvalas, 1988). Compound (I) was prepared in pure form according to the method described by Dong & Vennerstrom (2001). No traces of trimeric acetone peroxide were detected by 1H– and 13C NMR spectroscopy. The compound was recrystallized from acetone, yielding clear colourless crystals suitable for structural analysis.
We analysed several samples and, in agreement with earlier investigations (Groth, 1967a,b), were unable to find pure untwinned crystals. Many reflections show a typical splitting, and the conventional autoindexing routine in SMART (Siemens, 1993) failed to find a plausible unit cell. However, a twin analysis of nearly 1000 reflections with the program GEMINI (Sparks, 2000) revealed multiple twinning with at least three different crystal components for our sample. Polysynthetic or multiple twinning usually occurs with perpendicularly twinned crystals, where all of the twin interfaces are parallel to one another. The program GEMINI (Sparks, 2000) was used to generate an HKLF-5 file, including reflections of all three components, for refinement with SHELXL97 (Sheldrick, 1997). Although it was possible to solve the structure and to find all H atoms, the quality of the structure parameters were still relatively poor. For non-merohedral twins, some reflections exactly overlap, some partially overlap and others do not overlap at all. The relatively poor data quality arises because the integration program SAINT-Plus (Bruker, 1999) was not designed to integrate partially overlapped reflections accurately. In addition, it was impossible to obtain an orientation matrix with reasonable accuracy for the third minor component. For these reasons, we isolated the predominant component for an additional experiment. In this case, it was possible to obtain a substantial number of pure reflections from the major component only and the structure could be solved by traditional methods. All structure parameters given in this paper were obtained from this experiment. H-atom coordinates and Uiso values were refined; the C—H distances are in the range 0.93 (2)–0.99 (2) Å.
Data collection: SMART (Siemens, 1993); cell refinement: SAINT-Plus (Bruker, 1999); data reduction: SAINT-Plus; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: Diamond (Brandenburg, 2001); software used to prepare material for publication: SHELXL97.
3,3,6,6-tetramethyl-1,2,4,5-tetroxane
top
Crystal data top
| C6H12O4 | F(000) = 160 |
| Mr = 148.16 | Dx = 1.33 Mg m−3 |
| Monoclinic, P2/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yc | Cell parameters from 1547 reflections |
| a = 5.9194 (8) Å | θ = 3.4–31.3° |
| b = 5.9245 (8) Å | µ = 0.11 mm−1 |
| c = 10.5821 (14) Å | T = 208 K |
| β = 94.326 (3)° | Prism, colorless |
| V = 370.05 (9) Å3 | 0.4 × 0.1 × 0.1 mm |
| Z = 2 | |
Data collection top
Bruker SMART CCD area-detector
diffractometer | Rint = 0.037 |
| Radiation source: fine-focus sealed tube | θmax = 28.3°, θmin = 3.4° |
| ω scans | h = −7→7 |
| 2651 measured reflections | k = −5→7 |
| 919 independent reflections | l = −14→13 |
| 764 reflections with I > 2σ(I) | |
Refinement top
| Refinement on F2 | 0 restraints |
| Least-squares matrix: full | All H-atom parameters refined |
| R[F2 > 2σ(F2)] = 0.045 | w = 1/[σ2(Fo2) + (0.0481P)2 + 0.1358P]
where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.113 | (Δ/σ)max = 0.001 |
| S = 1.11 | Δρmax = 0.31 e Å−3 |
| 919 reflections | Δρmin = −0.22 e Å−3 |
| 70 parameters | |
Crystal data top
| C6H12O4 | V = 370.05 (9) Å3 |
| Mr = 148.16 | Z = 2 |
| Monoclinic, P2/c | Mo Kα radiation |
| a = 5.9194 (8) Å | µ = 0.11 mm−1 |
| b = 5.9245 (8) Å | T = 208 K |
| c = 10.5821 (14) Å | 0.4 × 0.1 × 0.1 mm |
| β = 94.326 (3)° | |
Data collection top
Bruker SMART CCD area-detector
diffractometer | 764 reflections with I > 2σ(I) |
| 2651 measured reflections | Rint = 0.037 |
| 919 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.045 | 0 restraints |
| wR(F2) = 0.113 | All H-atom parameters refined |
| S = 1.11 | Δρmax = 0.31 e Å−3 |
| 919 reflections | Δρmin = −0.22 e Å−3 |
| 70 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| O1 | 0.57499 (16) | 0.29345 (18) | 1.04397 (9) | 0.0273 (3) | |
| O2 | 0.29857 (17) | 0.56995 (17) | 1.04350 (9) | 0.0272 (3) | |
| C1 | 0.3386 (2) | 0.3369 (2) | 1.01527 (13) | 0.0251 (3) | |
| C2 | 0.2575 (3) | 0.2731 (3) | 0.88047 (14) | 0.0307 (4) | |
| C3 | 0.2210 (3) | 0.2074 (3) | 1.11473 (15) | 0.0330 (4) | |
| H2A | 0.332 (3) | 0.355 (4) | 0.8210 (19) | 0.043 (5)* | |
| H2B | 0.283 (3) | 0.109 (4) | 0.8691 (18) | 0.041 (5)* | |
| H2C | 0.098 (4) | 0.301 (4) | 0.8673 (19) | 0.043 (5)* | |
| H3A | 0.243 (3) | 0.051 (4) | 1.1050 (19) | 0.045 (6)* | |
| H3B | 0.059 (4) | 0.230 (4) | 1.106 (2) | 0.048 (6)* | |
| H3C | 0.276 (3) | 0.252 (3) | 1.1980 (18) | 0.033 (5)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| O1 | 0.0253 (5) | 0.0240 (6) | 0.0326 (5) | 0.0020 (4) | 0.0016 (4) | 0.0058 (4) |
| O2 | 0.0250 (5) | 0.0243 (6) | 0.0332 (5) | 0.0016 (4) | 0.0076 (4) | 0.0007 (4) |
| C1 | 0.0231 (7) | 0.0231 (7) | 0.0291 (7) | 0.0020 (6) | 0.0015 (5) | −0.0006 (5) |
| C2 | 0.0316 (8) | 0.0311 (8) | 0.0290 (7) | −0.0006 (7) | −0.0006 (6) | −0.0023 (6) |
| C3 | 0.0336 (8) | 0.0325 (9) | 0.0332 (8) | −0.0047 (7) | 0.0052 (6) | 0.0031 (7) |
Geometric parameters (Å, º) top
| O1—C1 | 1.4329 (17) | C2—H2B | 0.99 (2) |
| O1—O2i | 1.4753 (14) | C2—H2C | 0.96 (2) |
| O2—C1 | 1.4361 (18) | C3—H3A | 0.94 (2) |
| C1—C2 | 1.518 (2) | C3—H3B | 0.96 (2) |
| C1—C3 | 1.513 (2) | C3—H3C | 0.954 (19) |
| C2—H2A | 0.93 (2) | | |
| | | |
| C1—O1—O2i | 107.58 (9) | H2A—C2—H2B | 110.2 (17) |
| C1—O2—O1i | 107.31 (10) | C1—C2—H2C | 109.4 (12) |
| O1—C1—O2 | 107.60 (11) | H2A—C2—H2C | 108.8 (18) |
| O1—C1—C2 | 112.89 (12) | H2B—C2—H2C | 107.9 (17) |
| O1—C1—C3 | 104.74 (11) | C1—C3—H3A | 110.3 (12) |
| O2—C1—C2 | 112.91 (12) | C1—C3—H3B | 111.6 (13) |
| O2—C1—C3 | 104.54 (12) | H3A—C3—H3B | 105.6 (18) |
| C3—C1—C2 | 113.45 (13) | C1—C3—H3C | 111.0 (12) |
| C1—C2—H2A | 111.9 (13) | H3A—C3—H3C | 109.6 (17) |
| C1—C2—H2B | 108.6 (11) | H3B—C3—H3C | 108.6 (17) |
| | | |
| O2i—O1—C1—O2 | −64.66 (13) | O1i—O2—C1—C3 | 175.46 (10) |
| O2i—O1—C1—C3 | −175.50 (11) | O1i—O2—C1—C2 | −60.76 (14) |
| O2i—O1—C1—C2 | 60.60 (15) | C1—O1—O2i—C1i | 64.47 (13) |
| O1i—O2—C1—O1 | 64.48 (12) | C1—O2—O1i—C1i | −64.47 (13) |
| Symmetry code: (i) −x+1, −y+1, −z+2. |
Experimental details
| Crystal data |
| Chemical formula | C6H12O4 |
| Mr | 148.16 |
| Crystal system, space group | Monoclinic, P2/c |
| Temperature (K) | 208 |
| a, b, c (Å) | 5.9194 (8), 5.9245 (8), 10.5821 (14) |
| β (°) | 94.326 (3) |
| V (Å3) | 370.05 (9) |
| Z | 2 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.11 |
| Crystal size (mm) | 0.4 × 0.1 × 0.1 |
| |
| Data collection |
| Diffractometer | Bruker SMART CCD area-detector
diffractometer |
| Absorption correction | – |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 2651, 919, 764 |
| Rint | 0.037 |
| (sin θ/λ)max (Å−1) | 0.666 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.045, 0.113, 1.11 |
| No. of reflections | 919 |
| No. of parameters | 70 |
| H-atom treatment | All H-atom parameters refined |
| Δρmax, Δρmin (e Å−3) | 0.31, −0.22 |
Selected geometric parameters (Å, º) top| O1—C1 | 1.4329 (17) | C1—C2 | 1.518 (2) |
| O1—O2i | 1.4753 (14) | C1—C3 | 1.513 (2) |
| O2—C1 | 1.4361 (18) | | |
| | | |
| C1—O1—O2i | 107.58 (9) | O1—C1—C3 | 104.74 (11) |
| C1—O2—O1i | 107.31 (10) | O2—C1—C2 | 112.91 (12) |
| O1—C1—O2 | 107.60 (11) | O2—C1—C3 | 104.54 (12) |
| O1—C1—C2 | 112.89 (12) | C3—C1—C2 | 113.45 (13) |
| | | |
| O2i—O1—C1—O2 | −64.66 (13) | O1i—O2—C1—C3 | 175.46 (10) |
| O2i—O1—C1—C3 | −175.50 (11) | O1i—O2—C1—C2 | −60.76 (14) |
| O2i—O1—C1—C2 | 60.60 (15) | C1—O1—O2i—C1i | 64.47 (13) |
| O1i—O2—C1—O1 | 64.48 (12) | | |
| Symmetry code: (i) −x+1, −y+1, −z+2. |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2004/03/00/fg1722/fg1722fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2004/03/00/fg1722/fg1722fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2004/03/00/fg1722/fg1722fig3thm.gif)

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry



Because of the particular reactivity of the O—O bond, the synthesis and reactivity of cyclic peroxides has attracted many investigators (Schulz, 2000). Cyclic diperoxides have been used as efficient sources of radicals for the initiation of radical polymerization (Lockley et al., 2000), and the role of cyclic peroxides in the biosynthesis of prostacyclines and thromboxanes is well documented (van Dorp, 1979). Appropriately substituted 1,2,4-trioxanes and 1,2,4,5-tetroxanes display significant antimalarial activities (Jefford et al., 2000). As part of our general interest in cyclic peroxides and heterocyclic hydroperoxides, we have investigated the solid-state structure of 3,3,6,6-tetramethyl-1,2,4,5-tetraoxane (dimeric acetone peroxide), (I). Several thermochemical (Canizo & Cafferta, 1992; Murray et al., 1966; Wulz et al., 1970) and quantum-chemical (Diez & Jubert, 2000) data sets for (I) are available. Solvent effects on the inversion barrier of (I) have been studied (Aganov et al., 1970; Brune, et al., 1971), and a conformational analysis in the liquid phase suggested that the chair conformer is more stable than the twist form (Aganov et al., 1970; Murray et al., 1966).
Although (I) has been known for more than 100 years, surprisingly, the crystal structure has not been reported to date. This situation is unexpected given the great interest in this compound. In an initial attempt, Groth (1967a,b) reported structures of several dimeric peroxides, but (I) was left undetermined because of a twinning problem. On the other hand, the crystal structure of the similar copound dimeric 1,3-dibromoacetone peroxide, (II) (Schulz et al., 1967), and the sulfur homologue, 3,3,6,6-tetramethyl-S-tetrathione, (Me)2(C2S4)(Me)2, (IV) (Korp et al., 1981), are well known. The aim of the present work was to provide a complete structural analysis of (I), in order to support the findings obtained from density-functional theory, molecular-dynamics calculations and spectroscopic measurements.
Our analysis shows that (I) is located on an inversion centre (Fig. 1), with a chair conformation that was found to be the stable conformer indicated by liquid-phase experiments (Murray et al., 1966; Aganov et al., 1970) and quantum-chemical calculations (Diez & Jubert, 2000). Selected geometric parameters are given in Table 1, while complete crystallographic data are available as supplementary material. It is of interest to note that (IV) crystallizes in twist-boat conformation (Korp et al., 1981), with crystallographically imposed −4 symmetry.
The observed O—O and C—C distances differ only slightly from the calculated values of 1.46 and 1.53 Å, respectively, while the measured and calculated (1.43 Å) C—O distances are nearly identical (Diez & Jubert, 2000). It should be noted however, that the 'experimental values' for acetone peroxide discussed by Diez & Jubert (2000) are actually values reported for dimeric cyclohexanone peroxide, (III) (Groth, 1967a,b), and not dimeric acetone peroxide.
The observed C1—O1—O2i, O2—C1—C2, O1—C1—C3 and C3—C1—C2 angles [Table 1; symmetry code: (i)] are close to the calculated values 107.9, 112.5, 104.5 and 114.0°, respectively (Diez & Jubert, 2000). Interestingly, the O—O, and C—O bond lengths, the C—O—O angle and the C1—O1—O2—C2 torsion angle differ significantly from the corresponding values for (II) (1.45 and 1.46 Å, and 110 and 61°, respectively) but lie in the typical range for dialkyl peroxides and other cyclic peroxides (Matsugo & Saito, 1992). The geometry around artom C1 is a distorted tetrahedron, possibly as a result of a repulsive C—H···O interaction (Groth, 1967a,b).
The problem with earlier X-ray investigations of (I) has been the twinning of crystals (Groth, 1967a,b). If, for example, two different orientations of a molecule in a crystal lattice are energetically nearly equivalent, very often a symmetry change can be observed and sometimes it is not possible to prevent twinning even under idealized conditions e.g. low temperature, slow crystal growth, crystallization from different solvents etc.
In Fig. 2(a), a general view of the unit cell of (I) is given; Fig. 2(b) shows a perspective view along [001], to clarify packing effects and the local symmetry environment.
For our three-component twin, the twin rotation tool in GEMINI (Sparks, 2000) revealed integer indices in the reciprocal space around [001], the c* axis vertical to the c face of the crystal. The rotation angle is very close to 180 and 90° for the two additional crystal components (Fig. 3). Using just X-ray data, it is impossible to distinguish between reflection twinning and twofold rotation twinning. However, with the data of the major crystal component, it was possible to determine the space group and to solve the structure. This enables us to speculate about the nature of the twinning phenomenon.
A graphical analysis of the twin interface is illustrated in Fig. 3. For rotation around 180°, the twin interface can better be described as a mirror plane, because (I) was found to crystallize in a centrosymmetric space group. Interestingly, the structure of 3,6-diphenyl-1,2,4,5-tetraoxacyclohexane was also found to crystallize as a twin with this relatively common (001) plane (Groth, 1967a,b). In both cases – 90 and 180° rotation around the c* vector – the two axial (C2 and C2i) and two equatorial (C3 and C3i) methyl atoms of the chair-shaped acetone peroxide have a local environment that is nearly the same as that of all other molecules in the lattice. These methyl atoms always point towards the centre of the O1—O2i– or O2—O1i-bridge of the neighbouring molecules. This fact is easily derived from the packing diagram (Fig. 2 b), viewed along [001]. As a result of this comparable symmetry environment, the total energy of a twinned crystal should be very close to that of a pure untwined sample. Obviously, this would be the reason for the high affinity of (I) to crystallize as a multiple twin.