Aluminium germanium lithium, AlGeLi, crystallizes in two cubic dimorphs. The structure of the
F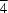
3
m form, already inferred from powder data, has been confirmed by both powder and single-crystal X-ray diffraction studies. The second dimorph, not previously identified, adopts a disordered centrosymmetric structure with space group
Fm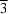 m
m.
Supporting information
Germanium powder (Strem chemicals, 99.999% pure) was used without purification; aluminium foil (Goodfellow, 99.999% pure, 0.5 mm) and lithium (ingot, Gogema, 99.94% pure, Mg free) were scraped to remove surface impurities. Alloys were prepared from the elements inside tantalum tubes, weld-sealed in argon atmosphere, and enclosed in stainless steel or silica jackets filled with argon to protect tantalum from oxidation at high temperatures. Mixtures of elements were heated to an appropriate temperature (about 1273 K) and kept at this temperature for several hours, during which they were shaken several times for good homogenization of the melts. The mixtures were then allowed to cool slowly to room temperature for crystal growth. Alloys were prepared at atomic ratios of 1:1:1 and 2:1:1 with the aim of obtaining the compounds LiAlGe and Li2AlGe previously cited in the literature. Crystals resulting from the two preparations were selected using a microscope inside a glove-box filled with purified argon. These crystals were inserted into thin-walled glass capillaries and then sealed to be used for checking singularity and crystal quality and for further X-ray investigations. The best diffracting single crystals were chosen for data collection at room temperature.
Data collection: CAD-4 Software (Enraf–Nonius, 1989) for (I); CrysAlis CCD (Oxford Diffraction, 2001) for (II). Cell refinement: CAD-4 Software for (I); CrysAlis RED (Oxford Diffraction, 2001) for (II). Data reduction: local program for (I); CrysAlis RED for (II). For both compounds, program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997). Molecular graphics: program (reference?) for (I); program (reference)? for (II). Software used to prepare material for publication: program (reference?) for (I); program (reference)? for (II).
(I) Lithium Aluminium Germanium
top
Crystal data top
| AlGeLi | Dx = 3.312 Mg m−3 |
| Mr = 106.51 | Mo Kα radiation, λ = 0.71070 Å |
| Cubic, F43m | Cell parameters from 25 reflections |
| Hall symbol: F -4 2 3 | θ = 5.9–20.9° |
| a = 5.9784 (9) Å | µ = 14.25 mm−1 |
| V = 213.67 (6) Å3 | T = 293 K |
| Z = 4 | Platelet, metallic dark grey |
| F(000) = 192 | 0.12 × 0.07 × 0.03 mm |
Data collection top
Enraf–Nonius CAD-4
diffractometer | 67 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.040 |
| Graphite monochromator | θmax = 34.5°, θmin = 5.9° |
| ω/θ scans | h = 0→9 |
Absorption correction: numerical
(numerical type provided by SHELX76; Sheldrick, 1976) | k = 0→9 |
| Tmin = 0.32, Tmax = 0.68 | l = −9→9 |
| 1083 measured reflections | 3 standard reflections every 100 reflections |
| 67 independent reflections | intensity decay: < 1% |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | w = 1/[σ2(Fo2) + (0.0094P)2 + 0.1227P]
where P = (Fo2 + 2Fc2)/3 |
| R[F2 > 2σ(F2)] = 0.011 | (Δ/σ)max < 0.001 |
| wR(F2) = 0.028 | Δρmax = 0.42 e Å−3 |
| S = 1.27 | Δρmin = −0.12 e Å−3 |
| 67 reflections | Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 6 parameters | Extinction coefficient: 0.030 (4) |
| 0 restraints | Absolute structure: Flack (1983), 28 Friedel pairs? |
| Primary atom site location: structure-invariant direct methods | Absolute structure parameter: −0.02 (4) |
Crystal data top
| AlGeLi | Z = 4 |
| Mr = 106.51 | Mo Kα radiation |
| Cubic, F43m | µ = 14.25 mm−1 |
| a = 5.9784 (9) Å | T = 293 K |
| V = 213.67 (6) Å3 | 0.12 × 0.07 × 0.03 mm |
Data collection top
Enraf–Nonius CAD-4
diffractometer | 67 reflections with I > 2σ(I) |
Absorption correction: numerical
(numerical type provided by SHELX76; Sheldrick, 1976) | Rint = 0.040 |
| Tmin = 0.32, Tmax = 0.68 | 3 standard reflections every 100 reflections |
| 1083 measured reflections | intensity decay: < 1% |
| 67 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.011 | 0 restraints |
| wR(F2) = 0.028 | Δρmax = 0.42 e Å−3 |
| S = 1.27 | Δρmin = −0.12 e Å−3 |
| 67 reflections | Absolute structure: Flack (1983), 28 Friedel pairs? |
| 6 parameters | Absolute structure parameter: −0.02 (4) |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Ge | 0.5000 | 0.5000 | 0.5000 | 0.0063 (2)* | |
| Li | 0.7500 | 0.7500 | 0.7500 | 0.016 (5)* | |
| Al | 0.2500 | 0.7500 | 0.7500 | 0.0073 (6)* | |
Geometric parameters (Å, º) top
| Ge—Ali | 2.5886 (4) | Li—Al | 2.9890 (5) |
| Ge—Li | 2.5886 (4) | | |
| | | |
| Ali—Ge—Alii | 109.5 | Ge—Li—Geiii | 109.5 |
| Li—Ge—Lii | 109.5 | | |
| Symmetry codes: (i) x, y−1/2, z−1/2; (ii) x+1/2, y−1/2, z; (iii) x+1/2, y+1/2, z. |
Crystal data top
| AlGeLi | Dx = 3.061 Mg m−3 |
| Mr = 106.51 | Mo Kα radiation, λ = 0.71073 Å |
| Cubic, Fm3m | Cell parameters from 1298 reflections |
| Hall symbol: -F 4 2 3 | θ = 5.8–34.6° |
| a = 6.1370 (8) Å | µ = 13.17 mm−1 |
| V = 231.14 (5) Å3 | T = 293 K |
| Z = 4 | Triangle, metallic dark grey |
| F(000) = 192 | 0.15 × 0.12 × 0.08 mm |
Data collection top
Oxford Diffraction Xcalibur CCD
diffractometer | 41 independent reflections |
| Radiation source: fine-focus sealed tube | 41 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.071 |
| ω scans | θmax = 34.6°, θmin = 5.8° |
Absorption correction: numerical
(CrysAlis RED; Oxford Diffraction, 2001; Clark & Reid, 1995) | h = −9→9 |
| Tmin = 0.20, Tmax = 0.41 | k = −9→9 |
| 1298 measured reflections | l = −9→9 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.038 | w = 1/[σ2(Fo2) + (0.0546P)2 + 2.038P]
where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.090 | (Δ/σ)max < 0.001 |
| S = 1.27 | Δρmax = 0.93 e Å−3 |
| 41 reflections | Δρmin = −0.72 e Å−3 |
| 4 parameters | Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 0 restraints | Extinction coefficient: 0.07 (2) |
Crystal data top
| AlGeLi | Z = 4 |
| Mr = 106.51 | Mo Kα radiation |
| Cubic, Fm3m | µ = 13.17 mm−1 |
| a = 6.1370 (8) Å | T = 293 K |
| V = 231.14 (5) Å3 | 0.15 × 0.12 × 0.08 mm |
Data collection top
Oxford Diffraction Xcalibur CCD
diffractometer | 41 independent reflections |
Absorption correction: numerical
(CrysAlis RED; Oxford Diffraction, 2001; Clark & Reid, 1995) | 41 reflections with I > 2σ(I) |
| Tmin = 0.20, Tmax = 0.41 | Rint = 0.071 |
| 1298 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.038 | 4 parameters |
| wR(F2) = 0.090 | 0 restraints |
| S = 1.27 | Δρmax = 0.93 e Å−3 |
| 41 reflections | Δρmin = −0.72 e Å−3 |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | Occ. (<1) |
| Ge | 0.0000 | 0.0000 | 0.0000 | 0.0231 (9)* | |
| Li1 | 0.2500 | 0.2500 | 0.2500 | 0.0264 (13)* | 0.50 |
| Al1 | 0.2500 | 0.2500 | 0.2500 | 0.0264 (13)* | 0.50 |
Geometric parameters (Å, º) top
| Ge—Li1/Al1 | 2.6574 (4) | Li1/Al1—Li1/Al1i | 3.0685 (5) |
| | | |
| Al1ii—Ge—Al1iii | 109.5 | Li1—Ge—Li1iv | 109.5 |
| Li1ii—Ge—Al1iii | 109.5 | | |
| Symmetry codes: (i) −x, −y+1/2, −z+1/2; (ii) −x, −y, −z; (iii) −x+1/2, −y+1/2, −z; (iv) x−1/2, y−1/2, z. |
Experimental details
| | (I) | (II) |
| Crystal data |
| Chemical formula | AlGeLi | AlGeLi |
| Mr | 106.51 | 106.51 |
| Crystal system, space group | Cubic, F43m | Cubic, Fm3m |
| Temperature (K) | 293 | 293 |
| a (Å) | 5.9784 (9) | 6.1370 (8) |
| V (Å3) | 213.67 (6) | 231.14 (5) |
| Z | 4 | 4 |
| Radiation type | Mo Kα | Mo Kα |
| µ (mm−1) | 14.25 | 13.17 |
| Crystal size (mm) | 0.12 × 0.07 × 0.03 | 0.15 × 0.12 × 0.08 |
| |
| Data collection |
| Diffractometer | Enraf–Nonius CAD-4
diffractometer | Oxford Diffraction Xcalibur CCD
diffractometer |
| Absorption correction | Numerical
(numerical type provided by SHELX76; Sheldrick, 1976) | Numerical
(CrysAlis RED; Oxford Diffraction, 2001; Clark & Reid, 1995) |
| Tmin, Tmax | 0.32, 0.68 | 0.20, 0.41 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 1083, 67, 67 | 1298, 41, 41 |
| Rint | 0.040 | 0.071 |
| (sin θ/λ)max (Å−1) | 0.798 | 0.798 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.011, 0.028, 1.27 | 0.038, 0.090, 1.27 |
| No. of reflections | 67 | 41 |
| No. of parameters | 6 | 4 |
| Δρmax, Δρmin (e Å−3) | 0.42, −0.12 | 0.93, −0.72 |
| Absolute structure | Flack (1983), 28 Friedel pairs? | ? |
| Absolute structure parameter | −0.02 (4) | ? |
Selected bond lengths (Å) for (I) top| Ge—Ali | 2.5886 (4) | Li—Al | 2.9890 (5) |
| Ge—Li | 2.5886 (4) | | |
| Symmetry code: (i) x, y−1/2, z−1/2. |
Selected bond lengths (Å) for (II) top| Ge—Li1/Al1 | 2.6574 (4) | Li1/Al1—Li1/Al1i | 3.0685 (5) |
| Symmetry code: (i) −x, −y+1/2, −z+1/2. |


 journal menu
journal menu inorganic compounds
inorganic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2005/05/00/fa1121/fa1121fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2005/05/00/fa1121/fa1121fig2thm.gif)




So far the literature related to the ternary system Li–Al–Ge has been very poor. On the basis of X-ray powder diffraction, a cubic non-centrosymmetric structure has been proposed for LiAlGe, with either ordered or disordered atomic arrangements (Nowotny & Holub, 1960; Bockelman & Schuster, 1974; Schuster et al., 1976). In all cases, the structure was depicted with one among four 43m sites empty. Bockelman & Schuster (1974) refer to the cubic compound Li2AlGe (a = 6.163 Å) without giving any more structural details. We decided to synthesize the compounds LiAlGe and Li2AlGe in order to define precisely their crystal structures.
The alloy prepared with the 1:1:1 stoichiometry leads to a homogeneous product, for which the X-ray powder diffraction pattern was perfectly indexed in a cubic cell [a = 5.9913 (1) Å] and further refined by the Rietveld technique in space group F43m to good agreement factors (Rp = 7.66% and RBragg = 4.92%). Meanwhile, the structure and the absolute configuration [Flack (1983) parameter = −0.01 (4)] were determined from good quality intensity data (Rint = 3.96%) collected from a single-crystal [a = 5.9784 (9) Å] on a Nonius single-point detector diffractometer. The structure refinement was carried out to R(F) = 1.13% [inversion of the configuration leads to an R(F) of 5.85%]. Our results confirm the ordered and non-centrosymmetric atomic arrangement for the LiAlGe polymorph (I). The atomic arrangement along the body diagonal of the cubic cell is vacancy (0,0,0) – Al (1/4,1/4,1/4) – Ge (1/2,1/2,1/2) – Li (3/4,3/4,3/4).
All attempts to prepare Li2AlGe were unsuccessful. Starting from the 2:1:1 stoichiometry, we obtained a material that displayed a two-phase X-ray powder pattern with the main component indexed in a cubic cell with an a parameter of 6.1528 (2) Å. The minor component lines were indexed using the DICVOL program (Boultif & Louer, 1991) in an I-centered tetragonal cell (a = 3.842 Å and c = 8.544 Å). A single-crystal could be extracted from this mixture and diffracted intensities were recorded on an Xcalibur CCD diffractometer. The statistical tests from SHELXS97 gave a strong evidence for centrosymmetry. The structure was first solved and refined with a disordered atomic arrangement in the space group Fm3m [a = 6.1370 (8) Å and R(F) = 3.76%].
Attempts to refine the structure in the other cubic symmetries F43m, F432 (m3m Laue symmetry), Fm3 and F23 (m3 Laue symmetry) gave no improvement. Since in some cases twinning can simulate centrosymmetry, we submitted the data to the twinning test server (Yeates, 1997); there was no hint of merohedral (perfect or partial) twinning. Nevertheless, we have tested various combinations of merohedral (mirror plane or twofold axis) and inversion twinning without success. Finally, the best solution was obtained in the Fm3m centrosymmetric space group with Li/Al atomic mixing at site 4b. The refinement of LiAlGe polymorph (II) is in accordance with the Al/Ge and Al/Li ratios obtained from SEM–EDX (scanning electron microcopy/energy-dispersive X-ray diffraction) and AAA (in full?) analyses.
In the non-centrosymmetric ordered form (I), the Ge atom is tetrahedrally surrounded by four Al and four Li atoms at 2.5886 (4) Å. In the centrosymmetric disordered polymorph (II), four Al and four Li atoms are statistically arranged at 2.6574 (4) Å at the apices of a cube around the Ge atom. Such statistical disorder in the Ge neighborhood might be responsible for the expansion of the unit cell.
In order to quantify how much such disorder can modify the unit-cell parameters, we have performed plane-wave DFT (denisty functional theory) calculations in the GGA/PBE approximation using the program CASTEP (Payne et al., 1992) distributed inside the Accelrys (2002) commercial package. Ultrasoft pseudopotentials were used for all atoms. The atomic disorder in (II) was modeled with the 2 × 2 × 2 supercell that retains the Fm3m symmetry. Geometry was optimized in the supercell with Ge atoms at sites a (multiplicity 4), b (4) and d (24), and Li and Al atoms at f (32) (x, x, x with refined x = 0.6233 and 0.8705, respectively). The optimized unit-cell parameter of 6.0921 Å for the subcell only deviates by 0.73% from the experimental one (a = 6.1370 Å). Comparatively, the geometry optimization of the F43m unit cell of (I) gave 6.0264 Å instead of 5.9784 Å (experimental, 0.80% deviation). The calculated total energies indicate that the non-centrosymmetric polymorph (I) is more stable than the centrosymmetric polymorph (II). The hypothetical compound Li2AlGe obtained by the Li-filling of vacancies in the cubic LiAlGe structures would display a cell parameter of 6.252 Å (Fm3m) or 6.406 Å (F43m) (Tillard et al., 2005).
In conclusion, we can assert that the compound LiAlGe exists in two cubic forms. The Al/Li substitutional disorder at site 4a (Fm3m centrosymmetric structure) is responsible for the 0.16 Å increase in the cell parameter with respect of the ordered F43m structure.