Buy article online - an online subscription or single-article purchase is required to access this article.
The crystal structure of H
3BO
3-3
T, a new trigonal polytype of orthoboric acid, consists of sheets of hydrogen-bonded B(OH)
3 molecules similar to those found in the triclinic structure of orthoboric acid, H
3BO
3-2
A. In each case, van der Waals forces connect the sheets. However, the stacking sequences of the sheets differ between the two polymorphs. In H
3BO
3-3
T (space group
P3
2), the sheets are stacked in the repeating sequence
ABC..., whereas in H
3BO
3-2
A (space group
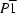
), the sheets are stacked in the repeating sequence
AB....
Supporting information
CCDC reference: 214361
Crystals of the title co,pound were prepared by mild hydrothermal techniques. The initial solution was prepared from NaBO2(H2O)2 (41 mg) and UO3 (29 mg; molar ratio 4:1) dissolved in ultrapure water (3 ml). The pH was adjusted to 0.5 using concentrated (15.4 mol/l) nitric acid. The resulting solution was placed in a 23 ml Teflon-lined Parr bomb, heated at 453 K for 48 h and then cooled to ambient temperature. No crystals had formed after heating, so the solution was placed in a fume hood to evaporate. After 2 d, well shaped pseudo-hexagonal crystals of H3BO3-2 A and small colorless crystals of H3BO3-3 T were recovered.
The positions of most O and B atoms were determined by direct methods. The remaining non-H atoms, and approximate positions of the H atoms, were located from subsequent difference Fourier syntheses. O—H bond distances were restrained to 0.96 Å, with standard uncertainties of 0.02 Å. There are no atoms heavier than O atoms in this structure, so that the absolute configuration cannot be determined reliably. Both absolute structures were tested and gave the same results. The 764 Friedel opposites were merged before final refinement, because the absolute structure parameter (Flack, 1983) was 4(3), and therefore unreliable. The twin law [010/100/00–1] was introduced in the final cycles of structure refinement, thus reducing the R(F) index from 0.089 to 0.044. The twin-component scale factor refined to 0.278 (2). The low standard uncertainties of the cell constants indicate the internal consistency of the measurements themselves, i.e. the precision of the measurements, not their accuracy.
Data collection: SMART (Bruker, 2001); cell refinement: SAINT-Plus (Bruker, 1999); data reduction: SAINT-Plus; program(s) used to solve structure: SHELXTL (Bruker, 1998); program(s) used to refine structure: SHELXTL; molecular graphics: ATOMS (Dowty, 2000); software used to prepare material for publication: SHELXTL and WinGX (Farrugia, 1999).
Crystal data top
| H3BO3 | Dx = 1.499 Mg m−3 |
| Mr = 61.83 | Mo Kα radiation, λ = 0.71073 Å |
| Trigonal, P32 | Cell parameters from 1623 reflections |
| Hall symbol: P 32 | θ = 3.3–33.8° |
| a = 7.0453 (4) Å | µ = 0.16 mm−1 |
| c = 9.5608 (7) Å | T = 297 K |
| V = 410.98 (4) Å3 | Triangular plate, colorless |
| Z = 6 | 0.14 × 0.08 × 0.02 mm |
| F(000) = 192 | |
Data collection top
Bruker APEX CCD area detector
diffractometer | 765 reflections with I > 2σ(I) |
| ω scans | Rint = 0.035 |
Absorption correction: empirical (using intensity measurements)
(XPREP; Bruker, 1997) | θmax = 34.4°, θmin = 2.1° |
| Tmin = 0.851, Tmax = 0.918 | h = −11→11 |
| 4615 measured reflections | k = −11→11 |
| 1143 independent reflections | l = −14→11 |
Refinement top
| Refinement on F2 | All H-atom parameters refined |
| Least-squares matrix: full | w = 1/[σ2(Fo2) + (0.0363P)2]
where P = (Fo2 + 2Fc2)/3 |
| R[F2 > 2σ(F2)] = 0.044 | (Δ/σ)max < 0.001 |
| wR(F2) = 0.088 | Δρmax = 0.28 e Å−3 |
| S = 1.00 | Δρmin = −0.25 e Å−3 |
| 1143 reflections | Extinction correction: SHELXTL (Bruker, 1998), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 99 parameters | Extinction coefficient: 0.051 (6) |
| 7 restraints | |
Crystal data top
| H3BO3 | Z = 6 |
| Mr = 61.83 | Mo Kα radiation |
| Trigonal, P32 | µ = 0.16 mm−1 |
| a = 7.0453 (4) Å | T = 297 K |
| c = 9.5608 (7) Å | 0.14 × 0.08 × 0.02 mm |
| V = 410.98 (4) Å3 | |
Data collection top
Bruker APEX CCD area detector
diffractometer | 1143 independent reflections |
Absorption correction: empirical (using intensity measurements)
(XPREP; Bruker, 1997) | 765 reflections with I > 2σ(I) |
| Tmin = 0.851, Tmax = 0.918 | Rint = 0.035 |
| 4615 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.044 | 7 restraints |
| wR(F2) = 0.088 | All H-atom parameters refined |
| S = 1.00 | Δρmax = 0.28 e Å−3 |
| 1143 reflections | Δρmin = −0.25 e Å−3 |
| 99 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| B1 | 0.1837 (9) | 0.6403 (10) | 0.2849 (7) | 0.0485 (17) | |
| B2 | 0.5180 (5) | 0.3069 (5) | 0.2964 (5) | 0.0277 (9) | |
| O1 | 0.0621 (6) | 0.7393 (7) | 0.2805 (3) | 0.0514 (8) | |
| O2 | 0.0864 (6) | 0.4195 (5) | 0.2808 (3) | 0.0467 (7) | |
| O3 | 0.4090 (5) | 0.7650 (6) | 0.2869 (3) | 0.0441 (8) | |
| O4 | 0.6437 (6) | 0.2103 (5) | 0.2929 (2) | 0.0413 (7) | |
| O5 | 0.2950 (6) | 0.1832 (6) | 0.2998 (3) | 0.0506 (8) | |
| O6 | 0.6136 (7) | 0.5294 (6) | 0.2904 (3) | 0.0464 (8) | |
| H1 | 0.133 (4) | 0.877 (3) | 0.316 (3) | 0.015 (7)* | |
| H2 | −0.057 (3) | 0.361 (5) | 0.311 (3) | 0.013 (7)* | |
| H3 | 0.483 (4) | 0.695 (4) | 0.311 (3) | 0.022 (7)* | |
| H4 | 0.565 (5) | 0.077 (4) | 0.333 (3) | 0.048 (10)* | |
| H5 | 0.243 (5) | 0.274 (4) | 0.324 (3) | 0.027 (8)* | |
| H6 | 0.755 (3) | 0.602 (4) | 0.318 (3) | 0.027 (8)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| B1 | 0.045 (3) | 0.056 (3) | 0.050 (3) | 0.029 (2) | −0.0005 (16) | −0.002 (2) |
| B2 | 0.0144 (12) | 0.0177 (14) | 0.053 (3) | 0.0093 (11) | 0.0007 (10) | 0.0015 (11) |
| O1 | 0.0246 (14) | 0.0384 (18) | 0.097 (2) | 0.0198 (14) | −0.0021 (15) | −0.0049 (16) |
| O2 | 0.0263 (15) | 0.0278 (14) | 0.0859 (19) | 0.0133 (13) | −0.0038 (16) | −0.0024 (14) |
| O3 | 0.0233 (12) | 0.0254 (14) | 0.082 (2) | 0.0108 (12) | −0.0006 (14) | −0.0032 (14) |
| O4 | 0.0279 (16) | 0.0249 (15) | 0.072 (2) | 0.0138 (14) | −0.0007 (13) | −0.0025 (12) |
| O5 | 0.0284 (14) | 0.0379 (18) | 0.0902 (19) | 0.0201 (13) | −0.0026 (15) | 0.0010 (16) |
| O6 | 0.0307 (19) | 0.0325 (15) | 0.0772 (18) | 0.0167 (13) | 0.0070 (15) | 0.0080 (13) |
Geometric parameters (Å, º) top
| B1—O1 | 1.349 (5) | O1—H1 | 0.91 (2) |
| B1—O2 | 1.350 (6) | O2—H2 | 0.93 (2) |
| B1—O3 | 1.377 (6) | O3—H3 | 0.91 (2) |
| B2—O4 | 1.361 (4) | O4—H4 | 0.90 (2) |
| B2—O5 | 1.364 (4) | O5—H5 | 0.91 (2) |
| B2—O6 | 1.363 (5) | O6—H6 | 0.91 (2) |
| | | |
| O1—B1—O2 | 120.4 (5) | O4—B2—O5 | 120.7 (3) |
| O1—B1—O3 | 119.9 (5) | O4—B2—O6 | 120.2 (3) |
| O2—B1—O3 | 119.6 (4) | O5—B2—O6 | 119.0 (2) |
| B1—O1—H1 | 113 (2) | B2—O4—H4 | 107 (2) |
| H1—O1—H6i | 111 (2) | H4—O4—H2iii | 120 (2) |
| B1—O1—H6i | 126 (1) | B2—O4—H2iii | 124 (1) |
| B1—O2—H2 | 108 (2) | B2—O5—H5 | 107 (2) |
| H2—O2—H5 | 120 (2) | H5—O5—H1iv | 124 (2) |
| B1—O2—H5 | 121 (1) | B2—O5—H1iv | 125.4 (8) |
| B1—O3—H3 | 117 (2) | B2—O6—H6 | 114 (2) |
| H3—O3—H4ii | 114 (2) | H6—O6—H3 | 112 (2) |
| B1—O3—H4ii | 123 (1) | B2—O6—H3 | 128 (1) |
| Symmetry codes: (i) x−1, y, z; (ii) x, y+1, z; (iii) x+1, y, z; (iv) x, y−1, z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O5ii | 0.91 (2) | 1.88 (2) | 2.715 (7) | 153 (2) |
| O2—H2···O4i | 0.93 (2) | 1.83 (2) | 2.705 (6) | 155 (2) |
| O3—H3···O6 | 0.91 (2) | 1.82 (2) | 2.688 (7) | 159 (3) |
| O4—H4···O3iv | 0.91 (2) | 1.95 (2) | 2.719 (5) | 141 (3) |
| O5—H5···O2 | 0.91 (2) | 1.89 (2) | 2.723 (6) | 151 (2) |
| O6—H6···O1iii | 0.91 (2) | 1.91 (2) | 2.740 (6) | 151 (3) |
| Symmetry codes: (i) x−1, y, z; (ii) x, y+1, z; (iii) x+1, y, z; (iv) x, y−1, z. |
Experimental details
| Crystal data |
| Chemical formula | H3BO3 |
| Mr | 61.83 |
| Crystal system, space group | Trigonal, P32 |
| Temperature (K) | 297 |
| a, c (Å) | 7.0453 (4), 9.5608 (7) |
| V (Å3) | 410.98 (4) |
| Z | 6 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.16 |
| Crystal size (mm) | 0.14 × 0.08 × 0.02 |
| |
| Data collection |
| Diffractometer | Bruker APEX CCD area detector
diffractometer |
| Absorption correction | Empirical (using intensity measurements)
(XPREP; Bruker, 1997) |
| Tmin, Tmax | 0.851, 0.918 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 4615, 1143, 765 |
| Rint | 0.035 |
| (sin θ/λ)max (Å−1) | 0.795 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.044, 0.088, 1.00 |
| No. of reflections | 1143 |
| No. of parameters | 99 |
| No. of restraints | 7 |
| H-atom treatment | All H-atom parameters refined |
| Δρmax, Δρmin (e Å−3) | 0.28, −0.25 |
Selected geometric parameters (Å, º) top| B1—O1 | 1.349 (5) | B2—O4 | 1.361 (4) |
| B1—O2 | 1.350 (6) | B2—O5 | 1.364 (4) |
| B1—O3 | 1.377 (6) | B2—O6 | 1.363 (5) |
| | | |
| O1—B1—O2 | 120.4 (5) | O4—B2—O5 | 120.7 (3) |
| O1—B1—O3 | 119.9 (5) | O4—B2—O6 | 120.2 (3) |
| O2—B1—O3 | 119.6 (4) | O5—B2—O6 | 119.0 (2) |
| B1—O1—H1 | 113 (2) | B2—O4—H4 | 107 (2) |
| B1—O2—H2 | 108 (2) | B2—O5—H5 | 107 (2) |
| B1—O3—H3 | 117 (2) | B2—O6—H6 | 114 (2) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O5i | 0.91 (2) | 1.88 (2) | 2.715 (7) | 153 (2) |
| O2—H2···O4ii | 0.93 (2) | 1.83 (2) | 2.705 (6) | 155 (2) |
| O3—H3···O6 | 0.91 (2) | 1.82 (2) | 2.688 (7) | 159 (3) |
| O4—H4···O3iii | 0.91 (2) | 1.95 (2) | 2.719 (5) | 141 (3) |
| O5—H5···O2 | 0.91 (2) | 1.89 (2) | 2.723 (6) | 151 (2) |
| O6—H6···O1iv | 0.91 (2) | 1.91 (2) | 2.740 (6) | 151 (3) |
| Symmetry codes: (i) x, y+1, z; (ii) x−1, y, z; (iii) x, y−1, z; (iv) x+1, y, z. |
Comparison of the unit-cell parameters in crystal structures of H3BO3-2 A and H3BO3-3 T top| Compound | a | b | c | α | β | γ |
| H3BO3-2 Aa | 7.039 (2) | 7.053 (2) | 6.578 (2) | 92.58 (2) | 101.17 (2) | 119.83 (2) |
| H3BO3-3 Tb | 7.0453 (4) | 7.0453 (4) | 9.5608 (7) | 90 | 90 | 120 |
| Notes: (a) Zachariasen (1954); (b) this work. |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu inorganic compounds
inorganic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2003/06/00/fa1014/fa1014fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2003/06/00/fa1014/fa1014fig2thm.gif)

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry



Structures of boric acids known to date include triclinic orthoboric acid, H3BO3-2 A (Zachariasen, 1934), its deuterated analog D3BO3 (Craven & Sabine, 1966), and three modifications of metaboric acid [first described by Kracek et al. (1934)], viz. orthorhombic α-HBO2 (Tazaki, 1940; Peters & Milberg, 1964), monoclinic β-HBO2 (Zachariasen, 1952, 1963b) and cubic γ-HBO2 (Zachariasen, 1963a). The crystal structures of the first three compounds contain sheets formed by B(OA3) (A = H and D) groups in orthoboric acid and by [B3O3(OH)3] groups in α-HBO2, with hydrogen bonding between these groups within a given sheet. In β-HBO2, two-thirds of the boron atoms have triangular coordination and one-third are tetrahedrally coordinated and linked into infinite chains, ∞1[B3O4(OH)(H2O)], arranged in sheets. In γ-HBO2, all B atoms have tetrahedral coordination and are linked into a three-dimensional framework, ∞3[BO(OH)] [see Lima-de-Faria et al. (1990) for formula nomenclature].
As mentioned by Craven & Sabine (1966), H3BO3-2 A was one of the first compounds with a hydrogen-bonded crystal structure to be examined by X-ray diffraction methods (Zachariasen, 1934) and, ever since, has been a `textbook' example. In the original work of Zachariasen (1934), an approximate crystal structure of H3BO3-2 A was reported. The (BO3)3− groups of nearly perfect C3 h symmetry were found to form pseudo-hexagonal sheets in which each (BO3)3− group has three adjacent groups. The H atoms were assumed to be positioned mid-way between the two nearest O atoms of adjacent (BO3)3− groups, but arguments favoring O—H···O hydrogen bonds in this structure were subsequently forwarded by Bernal & Megaw (1935). Studying disorder in the stacking of the sheets in thin microcrystals of H3BO3-2 A using electron diffraction, Cowley (1953) found that the H atoms are systematically displaced from the nearly collinear arrangement suggested by Zachariasen (1934). This model has not been verified in further X-ray (Zachariasen, 1954; Gajhede et al., 1986) and neutron (Craven & Sabine, 1966) diffraction experiments. Dorset (1992) performed electron-diffraction studies of thin microcrystals of H3BO3-2 A at low temperature (128 K and below) and determined that, as a result of a stacking disorder as discussed by Cowley (1953), microcrystals of orthoboric acid diffract as if single sheets were independent of one another. Amongst the most recent studies of triclinic orthoboric acid are the ab initio calculations of Zapol et al. (2000). Their theoretical results are in a good agreement with the experimental data, i.e. the binding energy of the molecules within a sheet is much higher than the interaction energy between the sheets, and this can explain the good lubricating properties of orthoboric acid (Erdemir et al., 1991).
This paper reports the crystal structure of a new polytype of orthoboric acid, which was isolated as an unexpected product during our attempts to synthesize new sodium uranyl borate compounds. As can be seen from Fig. 1, each of the two symmetrically independent B(OH)3 molecules has nearly perfect C3 h symmetry and is connected to three adjacent molecules by hydrogen bonding to form pseudo-hexagonal sheets, ∞2[B(OH)3], which are parallel to the (001) plane. Details are given in Tables 1 and 2. The average values of the B—O, O—H and O···H bond distances and O—B—O, B—O—H and O—H···O bond angles within a sheet are 1.36 (1), 0.91 (5) and 1.88 (5) Å, and 120.0 (9), 111 (5) and 152 (6)°, respectively. These values are similar to those of H3BO3-2 A (1.361, 0.88 and 1.85 Å, and 120.0, 112 and 171°; Zachariasen, 1954). The unit-cell parameters parallel to the (001) plane in both structures are also comparable (Table 3). Although the sheets in the crystal structure of H3BO3-3 T are of the same type as those in H3BO3-2 A, the stacking sequences of the sheets are different in these two structures. The sheets are stacked in the sequence ABC··· in H3BO3-3 T (Fig. 2a), whereas in H3BO3-2 A, the sheets are stacked in the sequence AB··· (Fig. 2 b). The distances between the planes of two adjacent sheets, calculated as half of d001 in H3BO3-2 A and one-third of d001 H3BO3-3 T, are 3.182 (2) and 3.1869 (2) Å, respectively, whereas the shortest interatomic distances between B and O atoms from neighboring sheets are 3.157 and 3.195 Å in H3BO3-2 A, and 3.172 (6) and 3.187 (8) Å in H3BO3-3 T. It appears that small tilts of the B(OH)3 molecules relative to the (001) plane in the crystal structure of H3BO3-3 T, almost of the same magnitude as those in H3BO3-2 A, are mainly due to weak interactions between B and O atoms, as mentioned by Zachariasen (1954) for H3BO3-2 A.