Hydrothermally prepared dilithium tin hexahydroxide crystallizes in the monoclinic system (space group
P2
1/
n), with the Sn atom at a site with
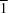
symmetry and all other atoms in general positions. The Sn coordination polyhedron is made up of six hydroxide groups. The Li atom is tetrahedrally coordinated by oxygen, with the tetrahedra sharing two corners and one edge with the adjacent Sn octahedra. Hydrogen bonds between the OH groups provide additional bonds in the framework.
Supporting information
The title compound was prepared by hydrothermal treatment of SnO (Fisher) and
LiOH (Aldrich) in a molar ratio of 1:3. The LiOH solution was prepared and
then mixed with SnO powder and stirred for 5 min. The cloudy liquid was
transferred to a Parr reactor and heated at 443 K for 3 d. The solution was
cooled and filtered, and white crystals of Li2Sn(OH)6 were separated
manually from the gray powder and dried at 330 K for 5 h.
All Sn and O atoms were located from the electron-density map. The Li atom and
remaining H atoms were located from the difference Fourier maps.
Data collection: SMART (Bruker, 1999); cell refinement: SAINT (Bruker, 1999); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 1990); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEP-3 for Windows (Farrugia, 1997) and ATOMS (Dowty, 1999); software used to prepare material for publication: SHELXL97.
Dilithium tin(VI) hexahydroxide
top
Crystal data top
| Li2Sn(OH)6 | F(000) = 220 |
| Mr = 234.62 | Dx = 3.024 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| a = 5.1640 (6) Å | Cell parameters from 929 reflections |
| b = 5.4013 (7) Å | θ = 8–57° |
| c = 9.2982 (11) Å | µ = 4.90 mm−1 |
| β = 96.596 (2)° | T = 293 K |
| V = 257.63 (5) Å3 | Irregular, colorless |
| Z = 2 | 0.12 × 0.08 × 0.06 mm |
Data collection top
Bruker SMART Apex CCD
diffractometer | 617 independent reflections |
| Radiation source: fine-focus sealed tube | 547 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.033 |
| ω scans | θmax = 28.8°, θmin = 4.3° |
Absorption correction: ψ-scan
(SADABS; Sheldrick, 1996) | h = −6→6 |
| Tmin = 0.65, Tmax = 0.75 | k = −5→7 |
| 1511 measured reflections | l = −11→12 |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.017 | All H-atom parameters refined |
| wR(F2) = 0.041 | w = 1/[σ2(Fo2) + (0.0167P)2]
where P = (Fo2 + 2Fc2)/3 |
| S = 1.06 | (Δ/σ)max < 0.001 |
| 617 reflections | Δρmax = 0.85 e Å−3 |
| 56 parameters | Δρmin = −0.50 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97 (Sheldrick, 1997), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0132 (18) |
Crystal data top
| Li2Sn(OH)6 | V = 257.63 (5) Å3 |
| Mr = 234.62 | Z = 2 |
| Monoclinic, P21/n | Mo Kα radiation |
| a = 5.1640 (6) Å | µ = 4.90 mm−1 |
| b = 5.4013 (7) Å | T = 293 K |
| c = 9.2982 (11) Å | 0.12 × 0.08 × 0.06 mm |
| β = 96.596 (2)° | |
Data collection top
Bruker SMART Apex CCD
diffractometer | 617 independent reflections |
Absorption correction: ψ-scan
(SADABS; Sheldrick, 1996) | 547 reflections with I > 2σ(I) |
| Tmin = 0.65, Tmax = 0.75 | Rint = 0.033 |
| 1511 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.017 | 0 restraints |
| wR(F2) = 0.041 | All H-atom parameters refined |
| S = 1.06 | Δρmax = 0.85 e Å−3 |
| 617 reflections | Δρmin = −0.50 e Å−3 |
| 56 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Sn | 0 | 1/2 | 1/2 | 0.00939 (13) | |
| O1 | 0.2180 (4) | 0.6013 (4) | 0.3381 (2) | 0.0141 (4) | |
| O2 | 0.2822 (4) | 0.2304 (4) | 0.5395 (2) | 0.0152 (4) | |
| O3 | 0.2221 (4) | 0.7324 (3) | 0.6360 (2) | 0.0146 (4) | |
| Li | 0.0964 (9) | 0.7626 (9) | 0.1589 (5) | 0.0192 (10) | |
| H1 | 0.318 (7) | 0.659 (7) | 0.367 (4) | 0.024 (11)* | |
| H2 | 0.246 (6) | 0.122 (6) | 0.547 (4) | 0.011 (10)* | |
| H3 | 0.183 (6) | 0.747 (6) | 0.707 (4) | 0.026 (10)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Sn | 0.00971 (17) | 0.00941 (17) | 0.00917 (17) | 0.00009 (8) | 0.00158 (9) | 0.00036 (8) |
| O1 | 0.0136 (10) | 0.0166 (11) | 0.0124 (9) | −0.0030 (9) | 0.0024 (8) | 0.0012 (8) |
| O2 | 0.0162 (10) | 0.0087 (10) | 0.0208 (11) | 0.0009 (8) | 0.0023 (8) | 0.0023 (9) |
| O3 | 0.0164 (10) | 0.0165 (10) | 0.0114 (10) | −0.0036 (7) | 0.0035 (7) | −0.0036 (8) |
| Li | 0.014 (2) | 0.023 (2) | 0.021 (2) | −0.0016 (19) | 0.0027 (18) | 0.002 (2) |
Geometric parameters (Å, º) top
| Sn—O3 | 2.0388 (19) | O1—Liii | 2.064 (5) |
| Sn—O3i | 2.0388 (19) | O2—Liii | 2.023 (5) |
| Sn—O1 | 2.055 (2) | O3—Liiii | 1.921 (5) |
| Sn—O1i | 2.055 (2) | Li—O1 | 1.920 (5) |
| Sn—O2 | 2.063 (2) | Li—O3iv | 1.921 (5) |
| Sn—O2i | 2.063 (2) | Li—O2v | 2.023 (5) |
| O1—Li | 1.920 (5) | Li—O1v | 2.064 (5) |
| | | |
| O3i—Sn—O3 | 180.0 | O1i—Sn—O2i | 83.30 (8) |
| O3i—Sn—O1 | 91.49 (8) | O2—Sn—O2i | 180.000 (1) |
| O3—Sn—O1 | 88.51 (8) | Li—O1—Sn | 127.19 (17) |
| O3i—Sn—O1i | 88.51 (8) | Li—O1—Liii | 121.24 (17) |
| O3—Sn—O1i | 91.49 (8) | Sn—O1—Liii | 92.68 (15) |
| O1—Sn—O1i | 180.000 (1) | Liii—O2—Sn | 93.63 (16) |
| O3i—Sn—O2 | 90.65 (8) | Liiii—O3—Sn | 124.39 (18) |
| O3—Sn—O2 | 89.35 (8) | O1—Li—O3iv | 109.1 (2) |
| O1—Sn—O2 | 83.30 (8) | O1—Li—O2v | 130.3 (3) |
| O1i—Sn—O2 | 96.70 (8) | O3iv—Li—O2v | 108.3 (2) |
| O3i—Sn—O2i | 89.35 (8) | O1—Li—O1v | 106.6 (2) |
| O3—Sn—O2i | 90.65 (8) | O3iv—Li—O1v | 116.7 (3) |
| O1—Sn—O2i | 96.70 (8) | O2v—Li—O1v | 84.05 (19) |
| Symmetry codes: (i) −x, −y+1, −z+1; (ii) −x+1/2, y−1/2, −z+1/2; (iii) x+1/2, −y+3/2, z+1/2; (iv) x−1/2, −y+3/2, z−1/2; (v) −x+1/2, y+1/2, −z+1/2. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O2vi | 0.64 (4) | 2.23 (4) | 2.847 (3) | 165 (4) |
| O2—H2···O3vii | 0.62 (3) | 2.27 (3) | 2.864 (3) | 161 (4) |
| O3—H3···O2viii | 0.72 (4) | 2.34 (4) | 3.020 (3) | 158 (3) |
| Symmetry codes: (vi) −x+1, −y+1, −z+1; (vii) x, y−1, z; (viii) −x+1/2, y+1/2, −z+3/2. |
Experimental details
| Crystal data |
| Chemical formula | Li2Sn(OH)6 |
| Mr | 234.62 |
| Crystal system, space group | Monoclinic, P21/n |
| Temperature (K) | 293 |
| a, b, c (Å) | 5.1640 (6), 5.4013 (7), 9.2982 (11) |
| β (°) | 96.596 (2) |
| V (Å3) | 257.63 (5) |
| Z | 2 |
| Radiation type | Mo Kα |
| µ (mm−1) | 4.90 |
| Crystal size (mm) | 0.12 × 0.08 × 0.06 |
| |
| Data collection |
| Diffractometer | Bruker SMART Apex CCD
diffractometer |
| Absorption correction | ψ-scan
(SADABS; Sheldrick, 1996) |
| Tmin, Tmax | 0.65, 0.75 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 1511, 617, 547 |
| Rint | 0.033 |
| (sin θ/λ)max (Å−1) | 0.678 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.017, 0.041, 1.06 |
| No. of reflections | 617 |
| No. of parameters | 56 |
| H-atom treatment | All H-atom parameters refined |
| Δρmax, Δρmin (e Å−3) | 0.85, −0.50 |
Selected bond lengths (Å) top| Sn—O3 | 2.0388 (19) | Li—O3i | 1.921 (5) |
| Sn—O1 | 2.055 (2) | Li—O2ii | 2.023 (5) |
| Sn—O2 | 2.063 (2) | Li—O1ii | 2.064 (5) |
| Li—O1 | 1.920 (5) | | |
| Symmetry codes: (i) x−1/2, −y+3/2, z−1/2; (ii) −x+1/2, y+1/2, −z+1/2. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O2iii | 0.64 (4) | 2.23 (4) | 2.847 (3) | 165 (4) |
| O2—H2···O3iv | 0.62 (3) | 2.27 (3) | 2.864 (3) | 161 (4) |
| O3—H3···O2v | 0.72 (4) | 2.34 (4) | 3.020 (3) | 158 (3) |
| Symmetry codes: (iii) −x+1, −y+1, −z+1; (iv) x, y−1, z; (v) −x+1/2, y+1/2, −z+3/2. |


 journal menu
journal menu inorganic compounds
inorganic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2001/03/00/br1309/br1309fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2001/03/00/br1309/br1309fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2001/03/00/br1309/br1309fig3thm.gif)




Recently, there has been much interest in tin oxide compounds as anodes in high energy density lithium batteries (Idota et al., 1997; Courtney & Dahn, 1997; Goward et al., 1999). In a search for new anode materials and in trying to understand the reduction mechanism of tin oxides, a systematic study of lithium tin oxides has been made by our group. This work presents the crystal structure of a new lithium tin hydroxide, Li2Sn(OH)6, resulting from this study.
The title structure is built up from slightly distorted Li tetrahedra and Sn octahedra (Fig. 1). Each Li tetrahedron shares two corners and one edge with three Sn octahedra (Fig. 2) to form a network. On the other hand, each Sn octahedron that lies on the center of symmetry shares two coplanar edges (O1—O2) and two corners (O3) to form a chain along the a axis. Two corners (O2) of the Sn octahedron are also shared with the Li tetrahedra even though they are already involved in the edge-sharing. The Sn—O2—Li-sharing links the chains into a three-dimensional network. Three hydrogen bonds (Table 2) between the OH groups form additional links in the network.
This structure is related to the hydrated lithium tin hydroxide compound, Li2[Sn(OH)6]·2H2O, reported previously by Reuter & Bargon (1997). It crystallizes in space group P21/n, with a = 5.023 (1), b = 6.923 (1) and c = 10.202 (3) Å, and β = 99.78 (1)°. In contrast with the title compound, there is no edge-sharing in the hydrate, where each Li tetrahedron shares three corners with Sn octahedra to form a three-dimensional framework. The forth corner is occupied by a water molecule (Fig. 3). The presence of these water groups increases the unit cell volume from the 257.6 Å3 in the title compound to 349.6 Å3 in the hydrate.
An Li2Sn(OH)6 material reported by Nakata & Toyooka (1997) also crystallizes in space group P21/n, with a = 10.2016 (5), b = 6.9246 (3) and c = 5.0255 (2) Å, and β=99.764 (3)°. Interestingly, the cell dimensions for this compound are essentially the same as those for the above-mentioned crystallohydrate (a and c axes are switched). This structure was solved using powder diffraction data and can be converted to the previous structure. However, while Sn and three O atoms convert to the same type of atoms, Li atoms convert to the O atoms of the water molecule. Thus the water molecule was misinterpreted as Li.