
The structure of the title compound has been determined using low-temperature (150 K) single-crystal X-ray and neutron diffraction data. Crystals adopt the uncommon space group
P4
2/
ncm and display a complex set of intermolecular interactions in which the water molecules play the crucial role: the water O-atom [O2(
w)] accepts two hydrogen bonds and both water H atoms act as bifurcated donors. A set of O—H

O hydrogen bonds is formed around the 4
2 axis comprising (
a) a cyclic tetrameric synthon involving four donor-H from two water molecules and two O(hydroxy) acceptors from two parent molecules, and (
b) short discrete O(hydroxy)—H
O2(
w) hydrogen bonds which link these tetramers along the
c axis. Four Br
Br interactions [3.708 (1) Å] form cyclic Br
4 tetramers around the
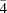
axis and are linked to the O—H
O system
via O2(
w)—H
Br bonds with H
Br = 2.995 (2) Å. Finally, the O—H
O system is further linked to the parent molecules
via C≡C
H
O2(
w) bonds of 2.354 (3) Å. The supramolecular structure of the title hydrate is compared with that of the non-hydrated parent molecule, which also forms cyclic O—H
O bonded tetrameric synthons, and with its (non-hydrated) tetrachloro analogue, which forms cyclic tetrameric Cl
4 synthons [Madhavi, Desiraju
et al. (2000
b).
Acta Cryst. B
56, 1063–1070].
Supporting information
CCDC references: 170351; 170352
Data collection: Bruker SMART for nmbrX. Cell refinement: Bruker SMART for nmbrX. Data reduction: Bruker SAINT for nmbrX. Program(s) used to solve structure: SHELXS97 (Sheldrick, 1990) for nlmbrN; SHELXS97 (Sheldrick, 1997) for nmbrX. For both compounds, program(s) used to refine structure: SHELXL97 (Sheldrick, 1997). Molecular graphics: SHELXL97 (Sheldrick, 1997) for nmbrX. Software used to prepare material for publication: SHELXL97 (Sheldrick, 1997) for nmbrX.
Crystal data top
| C10H4Br4O2·2H2O | F(000) = 348 |
| Mr = 511.80 | Dx = 2.429 Mg m−3 |
| Tetragonal, P42/ncm | Neutron radiation, λ = 0.5-5.0 Å |
| Hall symbol: -P 4ac 2ac | µ = 0.00 mm−1 |
| a = 12.6551 (18) Å | T = 150 K |
| c = 8.7340 (17) Å | Block, colourless |
| V = 1398.8 (4) Å3 | 3 × 2 × 2 mm |
| Z = 4 | |
Data collection top
`SXD'
diffractometer | 1654 reflections with I > 2σ(I) |
| Radiation source: ISIS spallation source | Rint = 0.000 |
| None monochromator | θmax = 62.6°, θmin = 2.3° |
| time–of–flight LAUE diffraction scans | h = 0→28 |
| 1654 measured reflections | k = 0→18 |
| 1654 independent reflections | l = 0→21 |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.061 | All H-atom parameters refined |
| wR(F2) = 0.082 | w = 1/[σ2(Fo2) + (0.P)2]
where P = (Fo2 + 2Fc2)/3 |
| S = 6.19 | (Δ/σ)max < 0.001 |
| 1654 reflections | Δρmax = 2.28 e Å−3 |
| 70 parameters | Δρmin = −1.40 e Å−3 |
| 0 restraints | Extinction correction: Becker-Coppens Lorentzian model |
| Primary atom site location: structure-invariant direct methods | |
Crystal data top
| C10H4Br4O2·2H2O | Z = 4 |
| Mr = 511.80 | Neutron radiation, λ = 0.5-5.0 Å |
| Tetragonal, P42/ncm | µ = 0.00 mm−1 |
| a = 12.6551 (18) Å | T = 150 K |
| c = 8.7340 (17) Å | 3 × 2 × 2 mm |
| V = 1398.8 (4) Å3 | |
Data collection top
`SXD'
diffractometer | 1654 reflections with I > 2σ(I) |
| 1654 measured reflections | Rint = 0.000 |
| 1654 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.061 | 0 restraints |
| wR(F2) = 0.082 | All H-atom parameters refined |
| S = 6.19 | Δρmax = 2.28 e Å−3 |
| 1654 reflections | Δρmin = −1.40 e Å−3 |
| 70 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Br1 | 0.58770 (6) | 0.23168 (6) | 0.11938 (8) | 0.02118 (13) | |
| C1 | 0.42464 (7) | 0.07536 (7) | 0.40923 (10) | 0.0273 (2) | |
| C2 | 0.43082 (6) | 0.06918 (6) | 0.27272 (8) | 0.01656 (16) | |
| C3 | 0.43183 (5) | 0.06817 (5) | 0.10514 (8) | 0.01116 (13) | |
| O1 | 0.35533 (6) | 0.14467 (6) | 0.05950 (10) | 0.01517 (18) | |
| C4 | 0.54095 (5) | 0.09963 (5) | 0.04716 (6) | 0.01210 (10) | |
| O2 | 0.34530 (8) | 0.15470 (8) | −0.24746 (12) | 0.0221 (2) | |
| H1A | 0.35202 (11) | 0.14798 (11) | −0.0533 (2) | 0.0266 (4) | |
| H2A | 0.36388 (17) | 0.22016 (16) | −0.2936 (2) | 0.0441 (4) | |
| H1 | 0.41968 (19) | 0.08032 (19) | 0.5303 (2) | 0.0579 (9) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Br1 | 0.0198 (3) | 0.0148 (3) | 0.0290 (3) | −0.0038 (3) | 0.0013 (2) | −0.0066 (2) |
| C1 | 0.0351 (4) | 0.0351 (4) | 0.0115 (3) | 0.0004 (5) | 0.0018 (2) | −0.0018 (2) |
| C2 | 0.0197 (3) | 0.0197 (3) | 0.0103 (2) | 0.0001 (3) | 0.00046 (18) | −0.00046 (18) |
| C3 | 0.0118 (2) | 0.0118 (2) | 0.0098 (2) | 0.0007 (3) | 0.00034 (16) | −0.00034 (16) |
| O1 | 0.0150 (3) | 0.0150 (3) | 0.0155 (3) | 0.0039 (4) | −0.0004 (2) | 0.0004 (2) |
| C4 | 0.0120 (2) | 0.0117 (2) | 0.01266 (17) | −0.0004 (2) | 0.00044 (18) | −0.00126 (18) |
| O2 | 0.0235 (4) | 0.0235 (4) | 0.0194 (4) | −0.0053 (5) | −0.0027 (3) | 0.0027 (3) |
| H1A | 0.0291 (6) | 0.0291 (6) | 0.0215 (6) | 0.0054 (8) | −0.0019 (5) | 0.0019 (5) |
| H2A | 0.0516 (12) | 0.0348 (9) | 0.0458 (9) | −0.0111 (9) | 0.0012 (8) | 0.0076 (7) |
| H1 | 0.0783 (14) | 0.0783 (14) | 0.0171 (8) | 0.0032 (18) | 0.0028 (7) | −0.0028 (7) |
Geometric parameters (Å, º) top
| Br1—C4 | 1.8817 (9) | C3—C4i | 1.5237 (7) |
| C1—C2 | 1.1974 (11) | C3—C4 | 1.5237 (7) |
| C1—H1 | 1.061 (2) | O1—H1A | 0.987 (2) |
| C2—C3 | 1.4637 (10) | C4—C4ii | 1.3347 (11) |
| C3—O1 | 1.4260 (13) | O2—H2A | 0.951 (2) |
| | | |
| C2—C1—H1 | 179.5 (3) | C2—C3—C4 | 109.75 (4) |
| C1—C2—C3 | 175.42 (11) | C4i—C3—C4 | 111.30 (7) |
| O1—C3—C2 | 105.52 (7) | C3—O1—H1A | 109.68 (14) |
| O1—C3—C4i | 110.18 (4) | C4ii—C4—C3 | 124.33 (3) |
| C2—C3—C4i | 109.75 (4) | C4ii—C4—Br1 | 121.74 (3) |
| O1—C3—C4 | 110.18 (4) | C3—C4—Br1 | 113.93 (5) |
| Symmetry codes: (i) −y+1/2, −x+1/2, z; (ii) y+1/2, x−1/2, −z. |
Crystal data top
| C10H8Br4O4 | Dx = 2.430 Mg m−3 |
| Mr = 511.80 | Mo Kα radiation, λ = 0.71073 Å |
| Tetragonal, P42/ncm | Cell parameters from 999 reflections |
| Hall symbol: -P 4ac 2ac | θ = 2.3–23.3° |
| a = 12.6551 (18) Å | µ = 11.51 mm−1 |
| c = 8.7340 (17) Å | T = 150 K |
| V = 1398.8 (4) Å3 | Block, colourless |
| Z = 4 | 0.4 × 0.3 × 0.3 mm |
| F(000) = 960 | |
Data collection top
Brucker SMART CCD
diffractometer | 539 independent reflections |
| Radiation source: fine-focus sealed tube | 501 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.030 |
| Detector resolution: not relevant pixels mm-1 | θmax = 23.3°, θmin = 2.3° |
| ω scans | h = −13→14 |
Absorption correction: multi-scan
multi-scan, using SADABS | k = −14→13 |
| Tmin = 0.644, Tmax = 1.000 | l = −8→9 |
| 6046 measured reflections | |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.020 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.053 | w = 1/[σ2(Fo2) + (0.0231P)2 + 2.4727P]
where P = (Fo2 + 2Fc2)/3 |
| S = 1.22 | (Δ/σ)max < 0.001 |
| 539 reflections | Δρmax = 0.44 e Å−3 |
| 58 parameters | Δρmin = −0.30 e Å−3 |
| 0 restraints | Extinction correction: SHELXL, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0039 (3) |
Crystal data top
| C10H8Br4O4 | Z = 4 |
| Mr = 511.80 | Mo Kα radiation |
| Tetragonal, P42/ncm | µ = 11.51 mm−1 |
| a = 12.6551 (18) Å | T = 150 K |
| c = 8.7340 (17) Å | 0.4 × 0.3 × 0.3 mm |
| V = 1398.8 (4) Å3 | |
Data collection top
Brucker SMART CCD
diffractometer | 539 independent reflections |
Absorption correction: multi-scan
multi-scan, using SADABS | 501 reflections with I > 2σ(I) |
| Tmin = 0.644, Tmax = 1.000 | Rint = 0.030 |
| 6046 measured reflections | θmax = 23.3° |
Refinement top
| R[F2 > 2σ(F2)] = 0.020 | 0 restraints |
| wR(F2) = 0.053 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.22 | Δρmax = 0.44 e Å−3 |
| 539 reflections | Δρmin = −0.30 e Å−3 |
| 58 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Br1 | 0.58779 (3) | 0.23210 (3) | 0.11967 (4) | 0.0307 (2) | |
| C1 | 0.4246 (3) | 0.0754 (3) | 0.4094 (7) | 0.0364 (14) | |
| H1 | 0.4196 | 0.0804 | 0.5154 | 0.06 (2)* | |
| C2 | 0.4310 (3) | 0.0690 (3) | 0.2751 (6) | 0.0272 (12) | |
| C3 | 0.4319 (3) | 0.0681 (3) | 0.1055 (5) | 0.0247 (11) | |
| O1 | 0.35500 (19) | 0.14500 (19) | 0.0611 (5) | 0.0267 (8) | |
| C4 | 0.5414 (3) | 0.0995 (3) | 0.0465 (4) | 0.0235 (8) | |
| O2 | 0.3458 (2) | 0.1542 (2) | −0.2463 (4) | 0.0322 (9) | |
| H2B | 0.357 (3) | 0.208 (3) | −0.281 (4) | 0.026 (11)* | |
| H1A | 0.348 (3) | 0.152 (3) | −0.018 (6) | 0.015 (16)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Br1 | 0.0304 (3) | 0.0257 (3) | 0.0360 (3) | −0.00322 (14) | 0.00125 (15) | −0.00608 (15) |
| C1 | 0.040 (2) | 0.040 (2) | 0.029 (4) | 0.004 (3) | 0.0004 (17) | −0.0004 (17) |
| C2 | 0.0269 (18) | 0.0269 (18) | 0.028 (3) | −0.001 (2) | 0.0008 (15) | −0.0008 (15) |
| C3 | 0.0256 (16) | 0.0256 (16) | 0.023 (3) | 0.002 (2) | 0.0005 (14) | −0.0005 (14) |
| O1 | 0.0302 (14) | 0.0302 (14) | 0.020 (2) | 0.0044 (15) | −0.0015 (12) | 0.0015 (12) |
| C4 | 0.0260 (19) | 0.0243 (19) | 0.0201 (18) | −0.0021 (14) | −0.0037 (13) | 0.0010 (13) |
| O2 | 0.0328 (14) | 0.0328 (14) | 0.031 (2) | −0.0054 (18) | −0.0058 (13) | 0.0058 (13) |
Geometric parameters (Å, º) top
| Br1—C4 | 1.889 (3) | C3—C4 | 1.531 (4) |
| C1—C2 | 1.178 (7) | O1—H2Bii | 2.32 (4) |
| C1—H1 | 0.9300 | O1—H1A | 0.70 (5) |
| C2—C3 | 1.482 (7) | C4—C4iii | 1.319 (7) |
| C3—O1 | 1.429 (6) | O2—H1A | 1.99 (5) |
| C3—C4i | 1.531 (4) | O2—H2B | 0.76 (3) |
| | | |
| C2—C1—H1 | 180.0 | C3—O1—H2Bii | 112.1 (10) |
| C1—C2—C3 | 175.0 (6) | C3—O1—H1A | 115 (4) |
| O1—C3—C2 | 105.1 (4) | H2Bii—O1—H1A | 120 (3) |
| O1—C3—C4i | 110.4 (3) | C4iii—C4—C3 | 124.56 (19) |
| C2—C3—C4i | 110.0 (3) | C4iii—C4—Br1 | 122.01 (10) |
| O1—C3—C4 | 110.4 (3) | C3—C4—Br1 | 113.4 (2) |
| C2—C3—C4 | 110.0 (3) | H1A—O2—H2B | 114 (3) |
| C4i—C3—C4 | 110.9 (4) | | |
| Symmetry codes: (i) −y+1/2, −x+1/2, z; (ii) y, −x+1/2, z+1/2; (iii) y+1/2, x−1/2, −z. |
Experimental details
| | (nlmbrN) | (nmbrX) |
| Crystal data |
| Chemical formula | C10H4Br4O2·2H2O | C10H8Br4O4 |
| Mr | 511.80 | 511.80 |
| Crystal system, space group | Tetragonal, P42/ncm | Tetragonal, P42/ncm |
| Temperature (K) | 150 | 150 |
| a, c (Å) | 12.6551 (18), 8.7340 (17) | 12.6551 (18), 8.7340 (17) |
| V (Å3) | 1398.8 (4) | 1398.8 (4) |
| Z | 4 | 4 |
| Radiation type | Neutron, λ = 0.5-5.0 Å | Mo Kα |
| µ (mm−1) | 0.00 | 11.51 |
| Crystal size (mm) | 3 × 2 × 2 | 0.4 × 0.3 × 0.3 |
| |
| Data collection |
| Diffractometer | `SXD'
diffractometer | Brucker SMART CCD
diffractometer |
| Absorption correction | – | Multi-scan
multi-scan, using SADABS |
| Tmin, Tmax | – | 0.644, 1.000 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 1654, 1654, 1654 | 6046, 539, 501 |
| Rint | 0.000 | 0.030 |
| θmax (°) | 62.6 | 23.3 |
| Distance from source to specimen (mm) | – | 0.555 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.061, 0.082, 6.19 | 0.020, 0.053, 1.22 |
| No. of reflections | 1654 | 539 |
| No. of parameters | 70 | 58 |
| H-atom treatment | All H-atom parameters refined | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 2.28, −1.40 | 0.44, −0.30 |


 journal menu
journal menu research papers
research papers










![[Figure 1]](http://journals.iucr.org/b/issues/2001/04/00/bm0041/bm0041fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/b/issues/2001/04/00/bm0041/bm0041fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/b/issues/2001/04/00/bm0041/bm0041fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/b/issues/2001/04/00/bm0041/bm0041fig4thm.gif)
![[Figure 5]](http://journals.iucr.org/b/issues/2001/04/00/bm0041/bm0041fig5thm.gif)
![[Figure 6]](http://journals.iucr.org/b/issues/2001/04/00/bm0041/bm0041fig6thm.gif)
![[Figure 7]](http://journals.iucr.org/b/issues/2001/04/00/bm0041/bm0041fig7thm.gif)



