

 journal menu
journal menu research papers
research papers









The crystal structures of piperazinium hexanoate-h11, 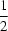 C4H12N2+2.C6H11O−2, and piperazinium hexanoate-d11, C4H12N2+2.C6D11O−2, have been determined from neutron diffraction data collected at 15 K. Nuclear anisotropic displacement parameters have been analyzed to obtain the internal molecular displacements of the H and D nuclei, given by 〈u2obs〉 − 〈u2ext〉 where 〈u2ext〉 is the contribution assuming all H/D to be carried rigidly on the vibrating molecular framework consisting of the heavier nuclei. In both crystal structures the cation ring is well fitted by the rigid-body model and the anion chain by a model with two rigid segments. In the piperazinium cations the corresponding protons in the two structures have about the same internal vibrational directions and magnitudes except for the two N—H protons, perhaps owing to differences in N—H
C4H12N2+2.C6H11O−2, and piperazinium hexanoate-d11, C4H12N2+2.C6D11O−2, have been determined from neutron diffraction data collected at 15 K. Nuclear anisotropic displacement parameters have been analyzed to obtain the internal molecular displacements of the H and D nuclei, given by 〈u2obs〉 − 〈u2ext〉 where 〈u2ext〉 is the contribution assuming all H/D to be carried rigidly on the vibrating molecular framework consisting of the heavier nuclei. In both crystal structures the cation ring is well fitted by the rigid-body model and the anion chain by a model with two rigid segments. In the piperazinium cations the corresponding protons in the two structures have about the same internal vibrational directions and magnitudes except for the two N—H protons, perhaps owing to differences in N—H O hydrogen bonding. The internal vibrations of corresponding H/D in the h11 and d11 anions have approximately the same vibrational directions. The internal mean-square displacements of the H nuclei are systematically greater than the values of the corresponding D nuclei by an average factor 1.7 (3). For both anions, normal-mode analyses have been carried out using the force fields derived from ab initio quantum-mechanical calculations with HF/3-21 G and HF/6-31G** basis sets. The values of the resultant H/D internal displacements for C—H(D) bond stretching and methylene out-of-plane vibrations are in good agreement with experiment. However, with either basis set, theory predicts methylene in-plane mean-square displacements significantly greater than the experimental values.
O hydrogen bonding. The internal vibrations of corresponding H/D in the h11 and d11 anions have approximately the same vibrational directions. The internal mean-square displacements of the H nuclei are systematically greater than the values of the corresponding D nuclei by an average factor 1.7 (3). For both anions, normal-mode analyses have been carried out using the force fields derived from ab initio quantum-mechanical calculations with HF/3-21 G and HF/6-31G** basis sets. The values of the resultant H/D internal displacements for C—H(D) bond stretching and methylene out-of-plane vibrations are in good agreement with experiment. However, with either basis set, theory predicts methylene in-plane mean-square displacements significantly greater than the experimental values.
O hydrogen bonding. The internal vibrations of corresponding H/D in the h11 and d11 anions have approximately the same vibrational directions. The internal mean-square displacements of the H nuclei are systematically greater than the values of the corresponding D nuclei by an average factor 1.7 (3). For both anions, normal-mode analyses have been carried out using the force fields derived from ab initio quantum-mechanical calculations with HF/3-21 G and HF/6-31G** basis sets. The values of the resultant H/D internal displacements for C—H(D) bond stretching and methylene out-of-plane vibrations are in good agreement with experiment. However, with either basis set, theory predicts methylene in-plane mean-square displacements significantly greater than the experimental values.



