Buy article online - an online subscription or single-article purchase is required to access this article.

trans-Dibromidotetrakis(1
H-pyrazole-κ
N3)manganese(II), [MnBr
2(C
3H
4N
2)
4], crystallizes in the
C2/
c space group with the Mn atom located on a centre of inversion. As a result, there is just one half-molecule in the asymmetric unit. Geometric parameters are in the usual ranges. The Mn centre is octahedrally coordinated by four pyrazole residues in the equatorial plane and by two bromide ligands in the axial positions. The molecular conformation is stabilized by N—H

Br hydrogen bonds. The structure of the title compound had already been described [Lumme & Lindell (1987).
J. Coord. Chem. 15, 383–392] in a different setting, with the Mn atoms located on inversion centres on Wyckoff position
d (
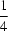
,
,
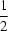
;
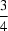
,
, 0;
,
,
;
,
, 0). In the conventional setting, however, the Mn atoms are situated on Wyckoff position
a (0, 0, 0; 0, 0,
;
,
, 0;
,
,
). In this special case, if the
c axis has the same length as the short diagonal of the
ac plane, the transformation from one setting into the other yields almost indistinguishable cell parameters, and the possibility of confusion arises. This setting ambiguity could be the reason why two structures in different settings might be taken as polymorphs even though they can easily be transformed. As a result of this, care should always be taken to use the conventional setting.
Supporting information
CCDC reference: 672400
A solution of dilithium scorpionate (30.1 mg, 0.059 mmol) in tetrahydrofuran (9 ml) was treated with [Mn(CO)5Br] (31.8 mg, 0.116 mmol). The solution was
separated via a cannula from the resulting precipitate. After removal
of the solvent, the residue was dissolved in benzene. After several weeks,
orange–yellow crystals of (II) suitable for X-ray diffraction were grown at
room temperature by slow evaporation of benzene.
H atoms were located in a difference map, but were positioned geometrically and
refined using a riding model, with fixed individual displacement parameters
[Uiso(H) = 1.2Ueq(C,N)] and with N—H = 0.88 Å and
C—H = 0.95 Å. The highest peak (2.75 e Å-3) in the final difference
electron-density map is 1.36 Å from Br1.
Data collection: X-AREA (Stoe & Cie, 2001); cell refinement: X-AREA (Stoe & Cie, 2001); data reduction: X-AREA (Stoe & Cie, 2001); program(s) used to solve structure: SHELXS97 (Sheldrick, 1990); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: PLATON (Spek, 2003) and XP in SHELXTL-Plus (Sheldrick, 1991); software used to prepare material for publication: SHELXL97 (Sheldrick, 1997).
Dibromidotetrakis(1
H-pyrazole-
κN3)manganese(II)
top
Crystal data top
| [MnBr2(C3H4N2)4] | F(000) = 956 |
| Mr = 487.09 | Dx = 1.841 Mg m−3 |
| Monoclinic, C2/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -C 2yc | Cell parameters from 5047 reflections |
| a = 14.430 (2) Å | θ = 3.5–25.7° |
| b = 9.4424 (8) Å | µ = 5.31 mm−1 |
| c = 14.725 (2) Å | T = 173 K |
| β = 118.864 (11)° | Block, light yellow |
| V = 1757.1 (4) Å3 | 0.17 × 0.16 × 0.14 mm |
| Z = 4 | |
Data collection top
Stoe IPDS II two-circle
diffractometer | 1644 independent reflections |
| Radiation source: fine-focus sealed tube | 1207 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.069 |
| ω scans | θmax = 25.6°, θmin = 3.5° |
Absorption correction: multi-scan
[MULABS (Spek, 2003; Blessing, 1995)] | h = −17→17 |
| Tmin = 0.41, Tmax = 0.47 | k = −11→11 |
| 8394 measured reflections | l = −17→17 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.066 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.169 | H-atom parameters constrained |
| S = 1.03 | w = 1/[σ2(Fo2) + (0.1049P)2]
where P = (Fo2 + 2Fc2)/3 |
| 1644 reflections | (Δ/σ)max < 0.001 |
| 106 parameters | Δρmax = 2.75 e Å−3 |
| 0 restraints | Δρmin = −0.83 e Å−3 |
Crystal data top
| [MnBr2(C3H4N2)4] | V = 1757.1 (4) Å3 |
| Mr = 487.09 | Z = 4 |
| Monoclinic, C2/c | Mo Kα radiation |
| a = 14.430 (2) Å | µ = 5.31 mm−1 |
| b = 9.4424 (8) Å | T = 173 K |
| c = 14.725 (2) Å | 0.17 × 0.16 × 0.14 mm |
| β = 118.864 (11)° | |
Data collection top
Stoe IPDS II two-circle
diffractometer | 1644 independent reflections |
Absorption correction: multi-scan
[MULABS (Spek, 2003; Blessing, 1995)] | 1207 reflections with I > 2σ(I) |
| Tmin = 0.41, Tmax = 0.47 | Rint = 0.069 |
| 8394 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.066 | 0 restraints |
| wR(F2) = 0.169 | H-atom parameters constrained |
| S = 1.03 | Δρmax = 2.75 e Å−3 |
| 1644 reflections | Δρmin = −0.83 e Å−3 |
| 106 parameters | |
Special details top
Experimental. ; |
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Mn1 | 0.5000 | 0.5000 | 0.5000 | 0.0217 (4) | |
| Br1 | 0.58177 (7) | 0.68530 (9) | 0.41305 (7) | 0.0283 (3) | |
| N1 | 0.5596 (5) | 0.3184 (8) | 0.4446 (5) | 0.0236 (14) | |
| N2 | 0.6167 (7) | 0.3385 (8) | 0.3954 (7) | 0.040 (2) | |
| H2 | 0.6356 | 0.4224 | 0.3840 | 0.048* | |
| C3 | 0.6418 (9) | 0.2159 (11) | 0.3657 (9) | 0.042 (2) | |
| H3 | 0.6817 | 0.2050 | 0.3303 | 0.051* | |
| C4 | 0.5982 (8) | 0.1098 (10) | 0.3968 (8) | 0.037 (2) | |
| H4 | 0.6017 | 0.0107 | 0.3877 | 0.045* | |
| C5 | 0.5476 (7) | 0.1789 (9) | 0.4447 (7) | 0.0263 (18) | |
| H5 | 0.5095 | 0.1321 | 0.4735 | 0.032* | |
| N11 | 0.3468 (5) | 0.4919 (7) | 0.3476 (5) | 0.0245 (14) | |
| N12 | 0.3291 (6) | 0.5611 (9) | 0.2605 (6) | 0.0317 (17) | |
| H12 | 0.3757 | 0.6168 | 0.2561 | 0.038* | |
| C13 | 0.2319 (7) | 0.5349 (11) | 0.1809 (8) | 0.037 (2) | |
| H13 | 0.2023 | 0.5723 | 0.1127 | 0.044* | |
| C14 | 0.1836 (7) | 0.4424 (12) | 0.2186 (9) | 0.046 (3) | |
| H14 | 0.1143 | 0.4039 | 0.1818 | 0.056* | |
| C15 | 0.2585 (6) | 0.4180 (9) | 0.3223 (7) | 0.0259 (18) | |
| H15 | 0.2481 | 0.3576 | 0.3682 | 0.031* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Mn1 | 0.0233 (8) | 0.0188 (9) | 0.0254 (9) | 0.0003 (6) | 0.0137 (7) | −0.0010 (7) |
| Br1 | 0.0344 (5) | 0.0217 (5) | 0.0350 (5) | −0.0016 (4) | 0.0216 (4) | 0.0011 (4) |
| N1 | 0.026 (3) | 0.023 (4) | 0.027 (3) | 0.001 (3) | 0.017 (3) | 0.000 (3) |
| N2 | 0.058 (5) | 0.024 (5) | 0.062 (5) | −0.002 (4) | 0.049 (5) | −0.001 (4) |
| C3 | 0.055 (6) | 0.034 (6) | 0.058 (6) | −0.001 (4) | 0.043 (5) | −0.007 (5) |
| C4 | 0.046 (5) | 0.019 (5) | 0.056 (6) | −0.003 (4) | 0.033 (5) | −0.012 (4) |
| C5 | 0.033 (4) | 0.016 (4) | 0.038 (5) | 0.001 (3) | 0.023 (4) | 0.002 (3) |
| N11 | 0.029 (3) | 0.020 (4) | 0.029 (4) | 0.000 (3) | 0.018 (3) | −0.003 (3) |
| N12 | 0.034 (4) | 0.032 (4) | 0.030 (4) | 0.001 (3) | 0.016 (3) | 0.001 (3) |
| C13 | 0.034 (5) | 0.035 (6) | 0.030 (5) | 0.006 (4) | 0.007 (4) | −0.006 (4) |
| C14 | 0.022 (4) | 0.041 (6) | 0.053 (7) | 0.000 (4) | −0.001 (4) | −0.015 (5) |
| C15 | 0.025 (4) | 0.017 (4) | 0.041 (5) | −0.002 (3) | 0.020 (4) | 0.000 (3) |
Geometric parameters (Å, º) top
| Mn1—N1 | 2.241 (7) | C4—C5 | 1.397 (13) |
| Mn1—N1i | 2.241 (7) | C4—H4 | 0.9500 |
| Mn1—N11i | 2.267 (7) | C5—H5 | 0.9500 |
| Mn1—N11 | 2.267 (7) | N11—C15 | 1.339 (10) |
| Mn1—Br1i | 2.7480 (9) | N11—N12 | 1.349 (10) |
| Mn1—Br1 | 2.7481 (9) | N12—C13 | 1.347 (12) |
| N1—C5 | 1.329 (11) | N12—H12 | 0.8800 |
| N1—N2 | 1.349 (10) | C13—C14 | 1.390 (17) |
| N2—C3 | 1.347 (13) | C13—H13 | 0.9500 |
| N2—H2 | 0.8800 | C14—C15 | 1.400 (14) |
| C3—C4 | 1.374 (15) | C14—H14 | 0.9500 |
| C3—H3 | 0.9500 | C15—H15 | 0.9500 |
| | | |
| N1—Mn1—N1i | 180.0 (3) | C4—C3—H3 | 126.9 |
| N1—Mn1—N11i | 91.4 (2) | C3—C4—C5 | 105.3 (8) |
| N1i—Mn1—N11i | 88.6 (2) | C3—C4—H4 | 127.4 |
| N1—Mn1—N11 | 88.6 (2) | C5—C4—H4 | 127.4 |
| N1i—Mn1—N11 | 91.4 (2) | N1—C5—C4 | 111.3 (7) |
| N11i—Mn1—N11 | 180.0 (3) | N1—C5—H5 | 124.4 |
| N1—Mn1—Br1i | 90.50 (18) | C4—C5—H5 | 124.4 |
| N1i—Mn1—Br1i | 89.50 (18) | C15—N11—N12 | 105.7 (7) |
| N11i—Mn1—Br1i | 89.59 (18) | C15—N11—Mn1 | 129.4 (6) |
| N11—Mn1—Br1i | 90.41 (18) | N12—N11—Mn1 | 124.9 (5) |
| N1—Mn1—Br1 | 89.50 (18) | C13—N12—N11 | 112.2 (8) |
| N1i—Mn1—Br1 | 90.50 (18) | C13—N12—H12 | 123.9 |
| N11i—Mn1—Br1 | 90.41 (18) | N11—N12—H12 | 123.9 |
| N11—Mn1—Br1 | 89.59 (18) | N12—C13—C14 | 106.3 (9) |
| Br1i—Mn1—Br1 | 180.0 | N12—C13—H13 | 126.9 |
| C5—N1—N2 | 104.7 (7) | C14—C13—H13 | 126.9 |
| C5—N1—Mn1 | 133.3 (5) | C13—C14—C15 | 105.5 (8) |
| N2—N1—Mn1 | 122.0 (5) | C13—C14—H14 | 127.2 |
| C3—N2—N1 | 112.6 (8) | C15—C14—H14 | 127.2 |
| C3—N2—H2 | 123.7 | N11—C15—C14 | 110.3 (8) |
| N1—N2—H2 | 123.7 | N11—C15—H15 | 124.8 |
| N2—C3—C4 | 106.2 (9) | C14—C15—H15 | 124.8 |
| N2—C3—H3 | 126.9 | | |
| | | |
| N11i—Mn1—N1—C5 | 96.4 (8) | N1—Mn1—N11—C15 | 89.7 (7) |
| N11—Mn1—N1—C5 | −83.6 (8) | N1i—Mn1—N11—C15 | −90.3 (7) |
| Br1i—Mn1—N1—C5 | 6.8 (7) | Br1i—Mn1—N11—C15 | −0.8 (7) |
| Br1—Mn1—N1—C5 | −173.2 (7) | Br1—Mn1—N11—C15 | 179.2 (7) |
| N11i—Mn1—N1—N2 | −87.2 (7) | N1—Mn1—N11—N12 | −87.8 (6) |
| N11—Mn1—N1—N2 | 92.8 (7) | N1i—Mn1—N11—N12 | 92.2 (6) |
| Br1i—Mn1—N1—N2 | −176.8 (7) | Br1i—Mn1—N11—N12 | −178.3 (6) |
| Br1—Mn1—N1—N2 | 3.2 (7) | Br1—Mn1—N11—N12 | 1.7 (6) |
| C5—N1—N2—C3 | −0.7 (11) | C15—N11—N12—C13 | 0.7 (10) |
| Mn1—N1—N2—C3 | −178.0 (7) | Mn1—N11—N12—C13 | 178.7 (6) |
| N1—N2—C3—C4 | 0.3 (13) | N11—N12—C13—C14 | −0.3 (11) |
| N2—C3—C4—C5 | 0.2 (13) | N12—C13—C14—C15 | −0.3 (11) |
| N2—N1—C5—C4 | 0.8 (10) | N12—N11—C15—C14 | −0.9 (10) |
| Mn1—N1—C5—C4 | 177.6 (6) | Mn1—N11—C15—C14 | −178.8 (6) |
| C3—C4—C5—N1 | −0.6 (12) | C13—C14—C15—N11 | 0.8 (11) |
| Symmetry code: (i) −x+1, −y+1, −z+1. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H2···Br1 | 0.88 | 2.70 | 3.342 (8) | 131 |
| N12—H12···Br1 | 0.88 | 2.82 | 3.437 (8) | 128 |
| N12—H12···Br1ii | 0.88 | 2.91 | 3.568 (8) | 133 |
| Symmetry code: (ii) −x+1, y, −z+1/2. |
Experimental details
| Crystal data |
| Chemical formula | [MnBr2(C3H4N2)4] |
| Mr | 487.09 |
| Crystal system, space group | Monoclinic, C2/c |
| Temperature (K) | 173 |
| a, b, c (Å) | 14.430 (2), 9.4424 (8), 14.725 (2) |
| β (°) | 118.864 (11) |
| V (Å3) | 1757.1 (4) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 5.31 |
| Crystal size (mm) | 0.17 × 0.16 × 0.14 |
| |
| Data collection |
| Diffractometer | Stoe IPDS II two-circle
diffractometer |
| Absorption correction | Multi-scan
[MULABS (Spek, 2003; Blessing, 1995)] |
| Tmin, Tmax | 0.41, 0.47 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 8394, 1644, 1207 |
| Rint | 0.069 |
| (sin θ/λ)max (Å−1) | 0.608 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.066, 0.169, 1.03 |
| No. of reflections | 1644 |
| No. of parameters | 106 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 2.75, −0.83 |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H2···Br1 | 0.88 | 2.70 | 3.342 (8) | 131.3 |
| N12—H12···Br1 | 0.88 | 2.82 | 3.437 (8) | 128.1 |
| N12—H12···Br1i | 0.88 | 2.91 | 3.568 (8) | 132.7 |
| Symmetry code: (i) −x+1, y, −z+1/2. |
Original and corrected (not necessary for 200, 250 and 298 K) values for the c
axis and β angle for biguanidinium bis(dinitramide) top | 85 K | 100 K | 150 K | 200 K | 250 K | 298 K |
| c original | 13.0023 | 13.0111 | 13.0302 | 13.0286 | 13.0265 | 13.0359 |
| c corrected | 12.8725 | 12.8987 | 12.9950 | | | |
| β original | 117.407 | 117.337 | 117.005 | 116.639 | 116.225 | 115.806 |
| β corrected | 116.270 | 116.354 | 116.699 | | | |
Original and corrected (not necessary for A) values for the c axis and β angle
for dichloridobis[3-(N-phenylamino)-3-methyl-2-butanone oxime]rhodium(III)
(Reference?) top | A | B |
| c original | 14.608 | 14.702 |
| c corrected | | 14.584 |
| β original | 106.57 | 108.23 |
| β corrected | | 106.77 |
Original and corrected (not necessary for II) values for the c axis and β
angle for
3αβ,4α-dihydro-4β,10-dimethyl-2-phenyl-1H,3H,5H-pyrrolo[3,4-b]carbazole-
1,3-dione (Reference?) top | Ia | IIa |
| c original | 23.729 | 23.663 |
| c corrected | 23.661 | |
| β original | 108.680 | 108.17 |
| β corrected | 108.188 | |
| (a) I and II refer to the structures I and II in the paper of Fischer et
al. (1995). |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu metal-organic compounds
metal-organic compounds










![[Figure 1]](http://journals.iucr.org/c/issues/2007/12/00/bg3059/bg3059fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2007/12/00/bg3059/bg3059fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2007/12/00/bg3059/bg3059fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/c/issues/2007/12/00/bg3059/bg3059fig4thm.gif)
![[Figure 5]](http://journals.iucr.org/c/issues/2007/12/00/bg3059/bg3059fig5thm.gif)

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry



Following the first synthesis of a scorpionate complex, considerable progress has been made towards extending this area of chemistry (Trofimenko, 1993). Our studies have shown that an important factor influencing the stability of scorpionates appears to be the degree of steric crowding around the boron centre (Bieller et al., 2004). Another reason for the deboronation of scorpionates may be the low difference in Lewis acidity of the metal centre in MX2 on one side and of the B centre in the corresponding borane of the pyrazolyl borate on the other side. The results of investigations in our group show that ditopic heteroscorpionates decompose in the presence of transition metal salts much more easily than monotopic homoscorpinates (Bieller et al., 2006).
It is interesting to note that the manganese complex [Mn(CO)5Br] can easily be transformed into MnII and Mn2(CO)10 in the presence of strong nucleophiles such as NaSSitBu3 or Na2PSitBu3 (Kückmann 2007; Lerner et al., 2005). In attempting to synthesize the 1,4-phenylene-bridged MnI scorpionate from the corresponding lithium scorpionate, (I), and [Mn(CO)5Br], we obtained the title compound, dibromido(tetrapyrazolato)manganese(II), (II), as a by-product (see reaction scheme).
A perspective view of compound (II) is shown in Fig. 1. The Mn centre is located on a centre of inversion. It is octahedrally coordinated by two Br atoms in a trans configuration occupying the axial positions and four pyrazole ligands in the equatorial plane. The pyrazole ligands are almost perpendicular [85.7 (31) and 88.9 (3)° for the rings containing atoms N1 and N11, respectively] to the equatorial plane consisting of the Mn centre and the four coordinating N atoms. The molecular conformation is stabilized by N—H···Br hydrogen bonds (Table 1).
In the monoclinic space group C2/c there are two different sets of inversion centres which are not equivalent, because they have a different environment. One set is on Wyckoff position a (0, 0, 0; 0, 0, 1/2; 1/2, 1/2, 0; 1/2, 1/2, 1/2) which can be transformed onto Wyckoff position b (0, 1/2, 0; 0, 1/2, 1/2; 1/2, 0, 0; 1/2, 0, 1/2) by an origin shift which is permitted in the monoclinic crystal system. Another set of inversion centres is on Wyckoff position c (1/4, 1/4, 0; 3/4, 1/4, 1/2; 3/4, 3/4, 0; 1/4, 3/4, 1/2) and Wyckoff position d (1/4, 1/4, 1/2; 3/4, 1/4, 0; 3/4, 3/4, 1/2; 1/4, 3/4, 0). The latter two sets can, similarly to a and b, be transformed into one another by a permitted origin shift. We have found for (II) that the Mn atoms are located on Wyckoff position a. This setting is hereinafter referred to as (IIa). The structure of the title compound has already been described (Lumme & Lindell, 1987) at 295 K in the same space group C2/c with cell parameters a = 14.208 (2), b = 9.454 (1) and c = 15.015 (3) Å, β = 118.68 (1)° and V = 1769.4 (5) Å3, but the Mn atoms are located on inversion centres of Wyckoff position d. This setting is hereinafter referred to as (IId).
At a first glance, the packing diagrams of the two settings (Figs. 2 and 3) look completely different. However, closer inspection of the packing patterns reveals that there are striking similarities. A superposition of both cells (Fig. 4) shows that they can be transformed into one another by the matrix (-1 0 0/0 - 1 0/ 1 0 1) and an additional origin shift of (1/4, 1/4, 0). The transformation of the symmetry elements is shown in Fig. 5. The conventional setting as given in International Tables for Crystallography Vol. A is shown in Fig. 5a. By applying the matrix (-1 0 0/0 - 1 0/ 1 0 1), which is equivalent to a twofold rotation about an axis perpendicular to the a axis, screw axes are interchanged with rotation axes and c-glide planes become n-glide planes and vice versa (Fig. 5b). The origin shift of 1/4 along a' (Fig. 5c) moves screw and rotation axes onto their correct positions. However, the height of the inversion centres and glide planes in the direction of the b axis is still not correct. This can be accomplished by an origin shift of 1/4 along b (Fig. 5d). Thus, in the special case where the c axis has almost the same length as the short diagonal of the ac plane, the transformation from one setting into one another yields almost indistinguishable cell parameters, and the possibility of confusion arises. Although refinement in each of these settings yields exactly the same results, only one of these settings is conventional. As a result of this, the cell parameters of (IId) should be transformed by the matrix (-1 0 0/0 - 1 0/ 1 0 1) into a = 14.208, b = 9.454 and c = 14.918 Å, and β = 117.99°, because the new c axis is shorter than the original one and the new β angle is smaller than the original one.
A search of the Cambridge Structural Database (Version 5.28, November 2006 plus three updates; Allen, 2002) for structures in the space group C2/c which are free of errors and not disordered yielded 29279 hits. For 713 of these, the condition that the length of the c axis does not deviate by more than 1% from the length of the shorter diagonal of the ac plane is fulfilled. Checking these 713 structures for whether a cell transformation by (-1 0 0/0 - 1 0/ 1 0 1) would give a shorter c axis and a smaller β angle revealed that 119 of these 713 would be better described in a different setting. If only one structure determinination is concerned, the results are exactly the same for both settings. However, among these 119 structure determinations, there are three which should be revised. Bolotina et al. (2003) compared six structures of biguanidinium bis(dinitramide) determined at different temperatures. However, for three of them, one setting was used and for the other three a different setting. Table 2 lists the original and corrected cell parameters. The original c axis at 150 K is the second longest (after that at 298 K). After transformation to a conventional setting, the length of the corrected c axis follows the expected trend of shortening when the temperature is lowered. Efe & Schlemper (1992) presented two structures of dichloro-bis(3-(N-phenylamino)-3-methyl-2-butanone oxime)rhodium(III), which are actually identical (Table 3). The same occurred with 3αβ,4α-dihydro-4β,10-dimethyl-2-phenyl-1H,3H,5H-pyrrolo[3,4-b]carbazole-1,3-dione (Fischer et al., 1995), who presented two polymorphs which are actually the same structure in a different setting (Table 4).
The nature of the problem described above is that some diffractometer programs might not calculate the correct conventional cell parameters and structure determination is performed in a non-conventional setting. A check for the conventional setting using the program PLATON (Spek, 2003) easily detects that a non-conventional setting has been used and lists the necessary transformation matrix. As far as just one single structure determination is concerned, it is just a question of sound crystallographic work. If, however, different determinations of the same structure are to be compared, care must be taken to use the same setting for all of these. And what is more obvious than using the conventional setting.