Unusually large and good-quality single crystals of the synthetic trioctahedral mica KFe
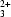
(Al
0.26Fe
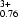
Si
3)O
10(OH)
2 [potassium triiron(II) aluminasilaferrate(III) decaoxide dihydroxide] have been grown hydrothermally. X-ray diffraction data measured at 270 and 100 K have been used to refine the crystal structure, including the positions of the H atoms. This synthetic mica is similar to annite, KFe
3AlSi
3O
10(OH)
2, and crystallizes with the same monoclinic
C2/
m symmetry. No phase transition has been observed down to 100 K. At low temperature, the ditrigonal distortion of the mica structure increases markedly, while the octahedral and tetrahedral bond lengths tend to decrease and increase, respectively. A detailed comparison of structural parameters in various Fe-rich micas is presented.
Supporting information
Single crystals of the title compound were synthesized under hydrothermal conditions using temperature-gradient experiments in 5 cm long Au tubes at 973 K under a hydrostatic pressure of 0.4 GPa. Stoichiometric amounts of K2CO3, Fe2O3, Al2O3 and SiO2 were carefully ground to a homogeneous fine-grained powder in an agate mortar under alcohol. This powder was the starting material for the synthesis. The oxygen fugacity during the experiment was determined by the high-pressure vessel and was close to the nickel/nickel-oxide solid-state buffer. A small number of unusually large mica single crystals with a nearly prismatic habit formed at the `cold' end of the Au capsule. This crystal shape is unusual as micas normally form thin flakes.
Several crystals were examined before the full data collection was carried out. Analysis of systematic absences confirmed the C-centred space group, identifying the polytype as 1-M. The composition of the tetrahedral site was determined by assuming an ideal Si content (3/4) and refining variable amounts of Al3+ (x) and Fe3+ (0.25 − x). The refined composition is close to that determined by microprobe analysis on fine-grained mica products of the same experiment (Redhammer et al., unpublished). Mellini et al. (1996) noted residual electron-density maxima located at b/3 from the original atom positions and interpreted them as due to faults in the stacking sequence along the [001] direction. No significant residual electron density was observed in the case of our crystals, including those with a large dimension along c. Structure refinements carried out on two additional crystals from the same synthesis batch yielded identical results to those presented here, within one standard uncertainty.
For both compounds, data collection: X-AREA (Stoe & Cie, 2002); cell refinement: X-AREA; data reduction: X-AREA; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: DIAMOND (Brandenburg & Berndt, 1999); software used to prepare material for publication: WinGX (Farrugia, 1999).
(I) potassium triiron(II) aluminasilaferrate(III) decaoxide dihydroxide
top
Crystal data top
| KFe3(Al0.26Fe0.76Si3)O10(OH)2 | F(000) = 519.7 |
| Mr = 533.82 | Dx = 3.425 Mg m−3 |
| Monoclinic, C2/m | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -C 2y | Cell parameters from 3729 reflections |
| a = 5.4208 (14) Å | θ = 2.1–32.3° |
| b = 9.3881 (17) Å | µ = 6.00 mm−1 |
| c = 10.330 (3) Å | T = 270 K |
| β = 100.06 (2)° | Cuboid, dark brown |
| V = 517.6 (2) Å3 | 0.17 × 0.14 × 0.10 mm |
| Z = 2 | |
Data collection top
Stoe IPDS 2
diffractometer | 814 reflections with I > 2σ(I) |
| Radiation source: sealed X-ray tube | Rint = 0.027 |
| rotation method scans | θmax = 32.1°, θmin = 2° |
Absorption correction: numerical
via equivalents (X-SHAPE and X-RED; Stoe & Cie 1996) | h = −7→8 |
| Tmin = 0.36, Tmax = 0.52 | k = −11→14 |
| 3927 measured reflections | l = −15→15 |
| 938 independent reflections | |
Refinement top
| Refinement on F2 | All H-atom parameters refined |
| Least-squares matrix: full | w = 1/[σ2(Fo2) + (0.0278P)2]
where P = (Fo2 + 2Fc2)/3 |
| R[F2 > 2σ(F2)] = 0.020 | (Δ/σ)max = 0.001 |
| wR(F2) = 0.049 | Δρmax = 0.40 e Å−3 |
| S = 1.10 | Δρmin = −0.50 e Å−3 |
| 938 reflections | Extinction correction: SHELXL97 (Sheldrick, 1997), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 59 parameters | Extinction coefficient: 0.0147 (8) |
| 1 restraint | |
Crystal data top
| KFe3(Al0.26Fe0.76Si3)O10(OH)2 | V = 517.6 (2) Å3 |
| Mr = 533.82 | Z = 2 |
| Monoclinic, C2/m | Mo Kα radiation |
| a = 5.4208 (14) Å | µ = 6.00 mm−1 |
| b = 9.3881 (17) Å | T = 270 K |
| c = 10.330 (3) Å | 0.17 × 0.14 × 0.10 mm |
| β = 100.06 (2)° | |
Data collection top
Stoe IPDS 2
diffractometer | 938 independent reflections |
Absorption correction: numerical
via equivalents (X-SHAPE and X-RED; Stoe & Cie 1996) | 814 reflections with I > 2σ(I) |
| Tmin = 0.36, Tmax = 0.52 | Rint = 0.027 |
| 3927 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.020 | 1 restraint |
| wR(F2) = 0.049 | All H-atom parameters refined |
| S = 1.10 | Δρmax = 0.40 e Å−3 |
| 938 reflections | Δρmin = −0.50 e Å−3 |
| 59 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | Occ. (<1) |
| Fe1 | 0.5 | 0.0 | 0.5 | 0.01006 (10) | |
| Fe2 | 1.0 | 0.16817 (2) | 0.5 | 0.01007 (9) | |
| Si | 0.57484 (6) | 0.16659 (3) | 0.22450 (3) | 0.01201 (12) | 0.75 |
| Fe3 | 0.57484 (6) | 0.16659 (3) | 0.22450 (3) | 0.01201 (12) | 0.189 (2) |
| Al | 0.57484 (6) | 0.16659 (3) | 0.22450 (3) | 0.01201 (12) | 0.061 (3) |
| K | 0.0 | 0.0 | 0.0 | 0.0370 (2) | |
| O1 | 0.8231 (2) | 0.23295 (15) | 0.16838 (11) | 0.0321 (3) | |
| O2 | 0.5218 (4) | 0.0 | 0.16822 (16) | 0.0322 (4) | |
| O3 | 0.62963 (18) | 0.16651 (9) | 0.38925 (10) | 0.01202 (19) | |
| O4 | 0.1309 (2) | 0.0 | 0.39564 (13) | 0.0121 (2) | |
| H | 0.106 (6) | 0.0 | 0.310 (4) | 0.035 (8)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Fe1 | 0.00842 (17) | 0.00838 (17) | 0.01370 (17) | 0.0 | 0.00281 (12) | 0.0 |
| Fe2 | 0.00802 (14) | 0.00859 (14) | 0.01370 (14) | 0.0 | 0.00221 (9) | 0.0 |
| Si | 0.01165 (17) | 0.01229 (17) | 0.01221 (17) | −0.00003 (11) | 0.00236 (11) | −0.00012 (11) |
| Fe3 | 0.01165 (17) | 0.01229 (17) | 0.01221 (17) | −0.00003 (11) | 0.00236 (11) | −0.00012 (11) |
| Al | 0.01165 (17) | 0.01229 (17) | 0.01221 (17) | −0.00003 (11) | 0.00236 (11) | −0.00012 (11) |
| K | 0.0432 (5) | 0.0448 (5) | 0.0225 (3) | 0.0 | 0.0040 (3) | 0.0 |
| O1 | 0.0334 (7) | 0.0431 (8) | 0.0198 (5) | −0.0082 (5) | 0.0053 (4) | −0.0017 (5) |
| O2 | 0.0466 (11) | 0.0298 (9) | 0.0186 (7) | 0.0 | 0.0012 (7) | 0.0 |
| O3 | 0.0104 (4) | 0.0114 (4) | 0.0147 (4) | 0.0001 (3) | 0.0035 (3) | 0.0002 (3) |
| O4 | 0.0115 (6) | 0.0129 (6) | 0.0119 (6) | 0.0 | 0.0022 (4) | 0.0 |
Geometric parameters (Å, º) top
| Fe1—O4 | 2.1004 (14) | Si—O1 | 1.6759 (13) |
| Fe1—O3i | 2.1277 (9) | Si—O1iii | 1.6765 (13) |
| Fe2—O4i | 2.1037 (9) | K—O2 | 3.050 (2) |
| Fe2—O3ii | 2.1195 (10) | K—O1iv | 3.0513 (14) |
| Fe2—O3 | 2.1301 (11) | K—O2iv | 3.364 (2) |
| Si—O2 | 1.6758 (7) | K—O1iii | 3.3637 (15) |
| Si—O3 | 1.6758 (11) | O4—H | 0.87 (4) |
| | | |
| O4i—Fe1—O3v | 95.65 (4) | O1x—K—O2xii | 129.93 (3) |
| O4—Fe1—O3v | 84.35 (4) | O1xi—K—O2xii | 50.07 (3) |
| O3v—Fe1—O3i | 94.56 (5) | O1xii—K—O2xii | 50.07 (3) |
| O3v—Fe1—O3 | 85.44 (5) | O2iv—K—O2xii | 180.00 (5) |
| O4i—Fe2—O4vi | 82.73 (5) | O2—K—O1iii | 50.06 (2) |
| O4i—Fe2—O3vii | 95.72 (4) | O2ix—K—O1iii | 129.94 (2) |
| O4vi—Fe2—O3vii | 178.14 (4) | O1iv—K—O1iii | 50.067 (17) |
| O3vii—Fe2—O3ii | 85.85 (5) | O1x—K—O1iii | 115.25 (4) |
| O3ii—Fe2—O3viii | 85.70 (4) | O1xi—K—O1iii | 64.75 (4) |
| O4vi—Fe2—O3viii | 84.21 (4) | O1xii—K—O1iii | 129.933 (17) |
| O4vi—Fe2—O3 | 95.16 (4) | O2iv—K—O1iii | 96.40 (3) |
| O3vii—Fe2—O3viii | 94.92 (4) | O2xii—K—O1iii | 83.60 (3) |
| O3viii—Fe2—O3 | 179.16 (5) | O2—K—O1xiii | 129.94 (2) |
| O2—Si—O3 | 109.94 (6) | O2ix—K—O1xiii | 50.06 (2) |
| O2—Si—O1 | 109.05 (8) | O1iv—K—O1xiii | 129.933 (17) |
| O3—Si—O1 | 109.94 (6) | O1x—K—O1xiii | 64.75 (4) |
| O2—Si—O1iii | 108.98 (8) | O1xi—K—O1xiii | 115.25 (4) |
| O3—Si—O1iii | 109.91 (5) | O1xii—K—O1xiii | 50.067 (17) |
| O1—Si—O1iii | 109.00 (4) | O2iv—K—O1xiii | 83.60 (3) |
| O2—K—O2ix | 180.00 (6) | O2xii—K—O1xiii | 96.40 (3) |
| O2—K—O1iv | 91.59 (4) | O1iii—K—O1xiii | 180.00 (4) |
| O2ix—K—O1iv | 88.41 (4) | O2—K—O1xiv | 50.06 (2) |
| O2—K—O1x | 91.59 (4) | O2ix—K—O1xiv | 129.94 (2) |
| O2ix—K—O1x | 88.41 (4) | O1iv—K—O1xiv | 115.25 (4) |
| O1iv—K—O1x | 91.57 (5) | O1x—K—O1xiv | 50.067 (17) |
| O2—K—O1xi | 88.41 (4) | O1xi—K—O1xiv | 129.933 (17) |
| O2ix—K—O1xi | 91.59 (4) | O1xii—K—O1xiv | 64.75 (4) |
| O1iv—K—O1xi | 88.43 (5) | O2iv—K—O1xiv | 96.40 (3) |
| O1x—K—O1xi | 180.00 (4) | O2xii—K—O1xiv | 83.60 (3) |
| O2—K—O1xii | 88.41 (4) | O1iii—K—O1xiv | 96.37 (5) |
| O2ix—K—O1xii | 91.59 (4) | O1xiii—K—O1xiv | 83.63 (5) |
| O1iv—K—O1xii | 180.00 (5) | O2—K—O1xv | 129.94 (2) |
| O1x—K—O1xii | 88.43 (5) | O2ix—K—O1xv | 50.06 (2) |
| O1xi—K—O1xii | 91.57 (5) | O1iv—K—O1xv | 64.75 (4) |
| O2—K—O2iv | 115.30 (5) | O1x—K—O1xv | 129.933 (17) |
| O2ix—K—O2iv | 64.70 (5) | O1xi—K—O1xv | 50.067 (17) |
| O1iv—K—O2iv | 50.07 (3) | O1xii—K—O1xv | 115.25 (4) |
| O1x—K—O2iv | 50.07 (3) | O2iv—K—O1xv | 83.60 (3) |
| O1xi—K—O2iv | 129.93 (3) | O2xii—K—O1xv | 96.40 (3) |
| O1xii—K—O2iv | 129.93 (3) | O1iii—K—O1xv | 83.63 (5) |
| O2—K—O2xii | 64.70 (5) | O1xiii—K—O1xv | 96.37 (5) |
| O2ix—K—O2xii | 115.30 (5) | O1xiv—K—O1xv | 180.00 (3) |
| O1iv—K—O2xii | 129.93 (3) | | |
| Symmetry codes: (i) −x+1, −y, −z+1; (ii) x+1/2, −y+1/2, z; (iii) x−1/2, −y+1/2, z; (iv) x−1, y, z; (v) −x+1, y, −z+1; (vi) x+1, y, z; (vii) −x+3/2, −y+1/2, −z+1; (viii) −x+2, y, −z+1; (ix) −x, −y, −z; (x) x−1, −y, z; (xi) −x+1, y, −z; (xii) −x+1, −y, −z; (xiii) −x+1/2, y−1/2, −z; (xiv) x−1/2, y−1/2, z; (xv) −x+1/2, −y+1/2, −z. |
(II) potassium triiron(II) aluminasilaferrate(III) decaoxide dihydroxide
top
Crystal data top
| KFe3(Al0.26Fe0.76Si3)O10(OH)2 | F(000) = 520.2 |
| Mr = 533.82 | Dx = 3.453 Mg m−3 |
| Monoclinic, C2/m | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -C 2y | Cell parameters from 4863 reflections |
| a = 5.4106 (13) Å | θ = 2.1–32.3° |
| b = 9.3709 (16) Å | µ = 6.07 mm−1 |
| c = 10.293 (3) Å | T = 100 K |
| β = 100.03 (2)° | Cuboid, dark brown |
| V = 513.9 (2) Å3 | 0.17 × 0.14 × 0.10 mm |
| Z = 2 | |
Data collection top
Stoe IPDS 2
diffractometer | 825 reflections with I > 2σ(I) |
| Radiation source: sealed X-ray tube | Rint = 0.028 |
| rotation method scans | θmax = 32.1°, θmin = 2.0° |
Absorption correction: numerical
via equivalents (X-SHAPE and X-RED; Stoe & Cie 1996) | h = −8→8 |
| Tmin = 0.36, Tmax = 0.52 | k = −11→14 |
| 4927 measured reflections | l = −15→15 |
| 934 independent reflections | |
Refinement top
| Refinement on F2 | All H-atom parameters refined |
| Least-squares matrix: full | w = 1/[σ2(Fo2) + (0.0298P)2 + 0.3868P]
where P = (Fo2 + 2Fc2)/3 |
| R[F2 > 2σ(F2)] = 0.019 | (Δ/σ)max < 0.001 |
| wR(F2) = 0.052 | Δρmax = 0.42 e Å−3 |
| S = 1.09 | Δρmin = −0.42 e Å−3 |
| 934 reflections | Extinction correction: SHELXL97 (Sheldrick, 1997), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 59 parameters | Extinction coefficient: 0.0090 (8) |
| 1 restraint | |
Crystal data top
| KFe3(Al0.26Fe0.76Si3)O10(OH)2 | V = 513.9 (2) Å3 |
| Mr = 533.82 | Z = 2 |
| Monoclinic, C2/m | Mo Kα radiation |
| a = 5.4106 (13) Å | µ = 6.07 mm−1 |
| b = 9.3709 (16) Å | T = 100 K |
| c = 10.293 (3) Å | 0.17 × 0.14 × 0.10 mm |
| β = 100.03 (2)° | |
Data collection top
Stoe IPDS 2
diffractometer | 934 independent reflections |
Absorption correction: numerical
via equivalents (X-SHAPE and X-RED; Stoe & Cie 1996) | 825 reflections with I > 2σ(I) |
| Tmin = 0.36, Tmax = 0.52 | Rint = 0.028 |
| 4927 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.019 | 1 restraint |
| wR(F2) = 0.052 | All H-atom parameters refined |
| S = 1.09 | Δρmax = 0.42 e Å−3 |
| 934 reflections | Δρmin = −0.42 e Å−3 |
| 59 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | Occ. (<1) |
| Fe1 | 0.5 | 0.0 | 0.5 | 0.00628 (10) | |
| Fe2 | 1.0 | 0.16807 (3) | 0.5 | 0.00630 (10) | |
| Si | 0.57461 (7) | 0.16660 (3) | 0.22365 (3) | 0.00888 (12) | 0.75 |
| Fe3 | 0.57461 (7) | 0.16660 (3) | 0.22365 (3) | 0.00888 (12) | 0.191 (3) |
| Al | 0.57461 (7) | 0.16660 (3) | 0.22365 (3) | 0.00888 (12) | 0.059 (3) |
| K | 0.0 | 0.0 | 0.0 | 0.01952 (16) | |
| O1 | 0.8284 (2) | 0.22740 (16) | 0.16760 (12) | 0.0253 (3) | |
| O2 | 0.5110 (4) | 0.0 | 0.16757 (17) | 0.0252 (4) | |
| O3 | 0.62975 (19) | 0.16648 (10) | 0.38892 (10) | 0.0089 (2) | |
| O4 | 0.1307 (3) | 0.0 | 0.39515 (13) | 0.0082 (3) | |
| H | 0.097 (7) | 0.0 | 0.303 (4) | 0.025 (9)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Fe1 | 0.00539 (17) | 0.00508 (17) | 0.00867 (17) | 0.0 | 0.00205 (12) | 0.0 |
| Fe2 | 0.00500 (14) | 0.00525 (14) | 0.00882 (14) | 0.0 | 0.00167 (9) | 0.0 |
| Si | 0.00882 (18) | 0.00905 (18) | 0.00893 (18) | −0.00015 (11) | 0.00198 (11) | −0.00008 (11) |
| Fe3 | 0.00882 (18) | 0.00905 (18) | 0.00893 (18) | −0.00015 (11) | 0.00198 (11) | −0.00008 (11) |
| Al | 0.00882 (18) | 0.00905 (18) | 0.00893 (18) | −0.00015 (11) | 0.00198 (11) | −0.00008 (11) |
| K | 0.0226 (3) | 0.0234 (3) | 0.0123 (3) | 0.0 | 0.0023 (2) | 0.0 |
| O1 | 0.0276 (7) | 0.0333 (8) | 0.0150 (5) | −0.0045 (5) | 0.0037 (4) | −0.0017 (5) |
| O2 | 0.0339 (10) | 0.0266 (9) | 0.0141 (7) | 0.0 | 0.0016 (7) | 0.0 |
| O3 | 0.0074 (5) | 0.0075 (5) | 0.0122 (4) | 0.0002 (3) | 0.0025 (3) | 0.0002 (3) |
| O4 | 0.0080 (6) | 0.0087 (6) | 0.0082 (6) | 0.0 | 0.0020 (4) | 0.0 |
Geometric parameters (Å, º) top
| Fe1—O4 | 2.0990 (15) | Si—O2 | 1.6792 (8) |
| Fe1—O3 | 2.1245 (10) | Si—O1iii | 1.6811 (14) |
| Fe2—O4i | 2.1003 (9) | K—O1iv | 2.9894 (15) |
| Fe2—O3ii | 2.1174 (10) | K—O2 | 2.993 (2) |
| Fe2—O3 | 2.1266 (12) | K—O2iv | 3.401 (2) |
| Si—O3 | 1.6751 (12) | K—O1iii | 3.4045 (16) |
| Si—O1 | 1.6790 (14) | O4—H | 0.94 (4) |
| | | |
| O4—Fe1—O3v | 95.60 (4) | O1xii—K—O2x | 129.79 (3) |
| O4vi—Fe1—O3v | 84.40 (4) | O2—K—O2x | 115.47 (6) |
| O3v—Fe1—O3 | 94.50 (6) | O2xiii—K—O2x | 64.53 (6) |
| O3vii—Fe1—O3 | 85.50 (6) | O2iv—K—O2x | 180.00 (3) |
| O4i—Fe2—O4vi | 82.84 (6) | O1iv—K—O1iii | 129.799 (17) |
| O4i—Fe2—O3viii | 178.21 (4) | O1x—K—O1iii | 50.201 (17) |
| O4vi—Fe2—O3viii | 95.66 (4) | O1xi—K—O1iii | 115.44 (4) |
| O3ii—Fe2—O3viii | 85.85 (6) | O1xii—K—O1iii | 64.56 (4) |
| O4vi—Fe2—O3 | 84.32 (5) | O2—K—O1iii | 50.19 (3) |
| O4i—Fe2—O3 | 95.07 (5) | O2xiii—K—O1iii | 129.81 (3) |
| O3viii—Fe2—O3 | 85.73 (4) | O2iv—K—O1iii | 82.73 (3) |
| O3ii—Fe2—O3 | 94.86 (4) | O2x—K—O1iii | 97.27 (3) |
| O3—Fe2—O3ix | 179.20 (5) | O1iv—K—O1xiv | 50.201 (17) |
| O3—Si—O1 | 109.72 (6) | O1x—K—O1xiv | 129.799 (17) |
| O3—Si—O2 | 109.76 (7) | O1xi—K—O1xiv | 64.56 (4) |
| O1—Si—O2 | 109.17 (8) | O1xii—K—O1xiv | 115.44 (4) |
| O3—Si—O1iii | 109.82 (6) | O2—K—O1xiv | 129.81 (3) |
| O1—Si—O1iii | 109.16 (5) | O2xiii—K—O1xiv | 50.19 (3) |
| O2—Si—O1iii | 109.19 (8) | O2iv—K—O1xiv | 97.27 (3) |
| O1iv—K—O1x | 180.00 (5) | O2x—K—O1xiv | 82.73 (3) |
| O1iv—K—O1xi | 89.07 (5) | O1iii—K—O1xiv | 180.00 (5) |
| O1x—K—O1xi | 90.93 (5) | O1iv—K—O1xv | 115.44 (4) |
| O1iv—K—O1xii | 90.93 (5) | O1x—K—O1xv | 64.56 (4) |
| O1x—K—O1xii | 89.07 (5) | O1xi—K—O1xv | 129.799 (17) |
| O1xi—K—O1xii | 180.00 (4) | O1xii—K—O1xv | 50.201 (17) |
| O1iv—K—O2 | 89.08 (4) | O2—K—O1xv | 129.81 (3) |
| O1x—K—O2 | 90.92 (4) | O2xiii—K—O1xv | 50.19 (3) |
| O1xi—K—O2 | 90.92 (4) | O2iv—K—O1xv | 97.27 (3) |
| O1xii—K—O2 | 89.08 (4) | O2x—K—O1xv | 82.73 (3) |
| O1iv—K—O2xiii | 90.92 (4) | O1iii—K—O1xv | 82.76 (5) |
| O1x—K—O2xiii | 89.08 (4) | O1xiv—K—O1xv | 97.24 (5) |
| O1xi—K—O2xiii | 89.08 (4) | O1iv—K—O1xvi | 64.56 (4) |
| O1xii—K—O2xiii | 90.92 (4) | O1x—K—O1xvi | 115.44 (4) |
| O2—K—O2xiii | 180.00 (6) | O1xi—K—O1xvi | 50.201 (17) |
| O1iv—K—O2iv | 50.21 (3) | O1xii—K—O1xvi | 129.799 (17) |
| O1x—K—O2iv | 129.79 (3) | O2—K—O1xvi | 50.19 (3) |
| O1xi—K—O2iv | 129.79 (3) | O2xiii—K—O1xvi | 129.81 (3) |
| O1xii—K—O2iv | 50.21 (3) | O2iv—K—O1xvi | 82.73 (3) |
| O2—K—O2iv | 64.53 (6) | O2x—K—O1xvi | 97.27 (3) |
| O2xiii—K—O2iv | 115.47 (6) | O1iii—K—O1xvi | 97.24 (5) |
| O1iv—K—O2x | 129.79 (3) | O1xiv—K—O1xvi | 82.76 (5) |
| O1x—K—O2x | 50.21 (3) | O1xv—K—O1xvi | 180.00 (5) |
| O1xi—K—O2x | 50.21 (3) | | |
| Symmetry codes: (i) x+1, y, z; (ii) x+1/2, −y+1/2, z; (iii) x−1/2, −y+1/2, z; (iv) −x+1, −y, −z; (v) x, −y, z; (vi) −x+1, −y, −z+1; (vii) −x+1, y, −z+1; (viii) −x+3/2, −y+1/2, −z+1; (ix) −x+2, y, −z+1; (x) x−1, y, z; (xi) x−1, −y, z; (xii) −x+1, y, −z; (xiii) −x, −y, −z; (xiv) −x+1/2, y−1/2, −z; (xv) −x+1/2, −y+1/2, −z; (xvi) x−1/2, y−1/2, z. |
Experimental details
| | (I) | (II) |
| Crystal data |
| Chemical formula | KFe3(Al0.26Fe0.76Si3)O10(OH)2 | KFe3(Al0.26Fe0.76Si3)O10(OH)2 |
| Mr | 533.82 | 533.82 |
| Crystal system, space group | Monoclinic, C2/m | Monoclinic, C2/m |
| Temperature (K) | 270 | 100 |
| a, b, c (Å) | 5.4208 (14), 9.3881 (17), 10.330 (3) | 5.4106 (13), 9.3709 (16), 10.293 (3) |
| β (°) | 100.06 (2) | 100.03 (2) |
| V (Å3) | 517.6 (2) | 513.9 (2) |
| Z | 2 | 2 |
| Radiation type | Mo Kα | Mo Kα |
| µ (mm−1) | 6.00 | 6.07 |
| Crystal size (mm) | 0.17 × 0.14 × 0.10 | 0.17 × 0.14 × 0.10 |
| |
| Data collection |
| Diffractometer | Stoe IPDS 2
diffractometer | Stoe IPDS 2
diffractometer |
| Absorption correction | Numerical
via equivalents (X-SHAPE and X-RED; Stoe & Cie 1996) | Numerical
via equivalents (X-SHAPE and X-RED; Stoe & Cie 1996) |
| Tmin, Tmax | 0.36, 0.52 | 0.36, 0.52 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 3927, 938, 814 | 4927, 934, 825 |
| Rint | 0.027 | 0.028 |
| (sin θ/λ)max (Å−1) | 0.748 | 0.748 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.020, 0.049, 1.10 | 0.019, 0.052, 1.09 |
| No. of reflections | 938 | 934 |
| No. of parameters | 59 | 59 |
| No. of restraints | 1 | 1 |
| H-atom treatment | All H-atom parameters refined | All H-atom parameters refined |
| Δρmax, Δρmin (e Å−3) | 0.40, −0.50 | 0.42, −0.42 |
Selected geometric parameters (Å, º) for (I) top| Fe1—O4 | 2.1004 (14) | Si—O1 | 1.6759 (13) |
| Fe1—O3i | 2.1277 (9) | Si—O1iii | 1.6765 (13) |
| Fe2—O4i | 2.1037 (9) | K—O2 | 3.050 (2) |
| Fe2—O3ii | 2.1195 (10) | K—O1iv | 3.0513 (14) |
| Fe2—O3 | 2.1301 (11) | K—O2iv | 3.364 (2) |
| Si—O2 | 1.6758 (7) | K—O1iii | 3.3637 (15) |
| Si—O3 | 1.6758 (11) | O4—H | 0.87 (4) |
| | | |
| O4i—Fe1—O3v | 95.65 (4) | O4vi—Fe2—O3 | 95.16 (4) |
| O4—Fe1—O3v | 84.35 (4) | O3vii—Fe2—O3viii | 94.92 (4) |
| O3v—Fe1—O3i | 94.56 (5) | O2—Si—O3 | 109.94 (6) |
| O3v—Fe1—O3 | 85.44 (5) | O2—Si—O1 | 109.05 (8) |
| O4i—Fe2—O4vi | 82.73 (5) | O3—Si—O1 | 109.94 (6) |
| O4i—Fe2—O3vii | 95.72 (4) | O2—Si—O1iii | 108.98 (8) |
| O3vii—Fe2—O3ii | 85.85 (5) | O3—Si—O1iii | 109.91 (5) |
| O3ii—Fe2—O3viii | 85.70 (4) | O1—Si—O1iii | 109.00 (4) |
| O4vi—Fe2—O3viii | 84.21 (4) | | |
| Symmetry codes: (i) −x+1, −y, −z+1; (ii) x+1/2, −y+1/2, z; (iii) x−1/2, −y+1/2, z; (iv) x−1, y, z; (v) −x+1, y, −z+1; (vi) x+1, y, z; (vii) −x+3/2, −y+1/2, −z+1; (viii) −x+2, y, −z+1. |
Selected geometric parameters (Å, º) for (II) top| Fe1—O4 | 2.0990 (15) | Si—O2 | 1.6792 (8) |
| Fe1—O3 | 2.1245 (10) | Si—O1iii | 1.6811 (14) |
| Fe2—O4i | 2.1003 (9) | K—O1iv | 2.9894 (15) |
| Fe2—O3ii | 2.1174 (10) | K—O2 | 2.993 (2) |
| Fe2—O3 | 2.1266 (12) | K—O2iv | 3.401 (2) |
| Si—O3 | 1.6751 (12) | K—O1iii | 3.4045 (16) |
| Si—O1 | 1.6790 (14) | O4—H | 0.94 (4) |
| | | |
| O4—Fe1—O3v | 95.60 (4) | O3viii—Fe2—O3 | 85.73 (4) |
| O4vi—Fe1—O3v | 84.40 (4) | O3ii—Fe2—O3 | 94.86 (4) |
| O3v—Fe1—O3 | 94.50 (6) | O3—Si—O1 | 109.72 (6) |
| O3vii—Fe1—O3 | 85.50 (6) | O3—Si—O2 | 109.76 (7) |
| O4i—Fe2—O4vi | 82.84 (6) | O1—Si—O2 | 109.17 (8) |
| O4vi—Fe2—O3viii | 95.66 (4) | O3—Si—O1iii | 109.82 (6) |
| O3ii—Fe2—O3viii | 85.85 (6) | O1—Si—O1iii | 109.16 (5) |
| O4vi—Fe2—O3 | 84.32 (5) | O2—Si—O1iii | 109.19 (8) |
| O4i—Fe2—O3 | 95.07 (5) | | |
| Symmetry codes: (i) x+1, y, z; (ii) x+1/2, −y+1/2, z; (iii) x−1/2, −y+1/2, z; (iv) −x+1, −y, −z; (v) x, −y, z; (vi) −x+1, −y, −z+1; (vii) −x+1, y, −z+1; (viii) −x+3/2, −y+1/2, −z+1. |
Structural parameters for selected Fe-bearing trioctahedral 1M micas. top| Sample | Anna | TFAb | TFAc | Rb-TFAd | Cs-TFAe |
| M1 site: | | | | | |
| Volume (Å3) | 12.63 (3) | 12.50 (2) | 12.46 (2) | 12.56 (6) | 12.67 (5) |
| BLDf (%) | 0.21 | 0.57 | 0.53 | 0.52 | 0.46 |
| ELDg (%) | 4.17 | 4.58 | 4.52 | 5.14 | 5.33 |
| OAVh (°) | 26.37 | 30.81 | 30.16 | 41.71 | 42.27 |
| ψi (°) | 58.28 | 58.57 | 58.53 | 59.23 | 59.29 |
| eu/esj | 1.089 | 1.097 | 1.096 | 1.109 | 1.114 |
| <M1-O> | 2.129 (1) | 2.119 (1) | 2.116 (1) | 2.126 (4) | 2.134 (5) |
| M2 site: | | | | | |
| Volume (Å3) | 12.56 (3) | 12.49 (2) | 12.44 (2) | 12.56 (3) | 12.61 (9) |
| BLDf (%) | 0.38 | 0.44 | 0.46 | 0.41 | 0.55 |
| ELDg (%) | 4.03 | 4.44 | 4.38 | 5.24 | 5.17 |
| OAVh (°) | 25.52 | 31.01 | 30.21 | 42.12 | 41.54 |
| ψi (°) | 58.20 | 58.55 | 58.51 | 59.23 | 59.19 |
| eu/esj | 1.087 | 1.096 | 1.095 | 1.114 | 1.113 |
| toct (Å) | 2.238 | 2.210 | 2.210 | 2.175 | 2.180 |
| <M2-O> | 2.124 (1) | 2.118 (1) | 2.115 | 2.126 (4) | 2.128 (5) |
| T site: | | | | | |
| Volume (Å3) | 2.36 (1) | 2.42 (1) | 2.43 (1) | 2.46 (1) | 2.47 (2) |
| BLDf (%) | 0.14 | 0.02 | 0.10 | 0.23 | 0.28 |
| TAVk (°) | 0.83 | 0.26 | 0.11 | 0.34 | 1.44 |
| <T-O> | 1.668 (1) | 1.676 (1) | 1.679 (1) | 1.687 (4) | 1.688 (4) |
| τ (°)l | 110.29 | 109.62 | 109.17 | 110.00 | 110.52 |
| α (°)m | 2.48 | 6.74 | 8.90 | 2.26 | 0.16 |
| Interlayer site: | | | | | |
| K-Oinner | 3.138 (1) | 3.051 (2) | 2.991 (2) | 3.227 (6) | 3.359 (6) |
| K-Oouter | 3.255 (1) | 3.364 (2) | 3.403 (2) | 3.330 (5) | 3.372 (6) |
| Δ(outer - inner) | 0.133 | 0.313 | 0.412 | 0.103 | 0.013 |
| Notes: (a) natural annite (Redhammer & Roth, 2002). (b) tetraferriannite at 270 K (this work). (c) tetraferriannite at 100 K (this work). (d) Rb-tetraferriannite (Comodi et al., 2003). (e) Cs-tetraferriannite (Comodi et al., 1999). (f) bond-length distortion BLD = (100/n)Σi=1n[{(X-O)i-(<x-O>)}/(<X-O>)], with n = number of bonds, (X-O)i = central cation-to-oxygen length and <X-O> = average cation-oxygen bond length (Renner & Lehmann, 1986). (g) edge-length distortion ELD = (100/n)Σi=1n[{(O-O)i-(<O-O>)}/(<O-O>)], with n = number of edges, (O-O)i = polyhedron edge length and <O-O> = average polyhedron edge length (Renner & Lehmann, 1986) (h) octahedral angle variance OAV = Σi=1n(Θi-90)2/11 (Robinson et al., 1971). (i) octahedral flattening angle ψ: cosψ = toct/2*doct, with doct = average M-O distance and toct = octahedral sheet thickness toct = 2[0.5-{2(zO3-zO4)/3}]c sinβ. (j) unshared edge eu/shared edge es (Toraya, 1981). (k) tetrahedral angle variance TAV = Σi=1n(Θi-109.47)2/5 (Robinson et al., 1971). (l) τ = mean of the three Obasal-T-Oapex angles. (m) α = ditrigonal distortion of the tetrahedral sheet with tanα = 4 × 31/2(0.25 − yO1) |


 journal menu
journal menu inorganic compounds
inorganic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2004/04/00/bc1034/bc1034fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2004/04/00/bc1034/bc1034fig2thm.gif)




Trioctahedral micas, XY3Z4O10(OH)2 (where X is an interlayer cation, Y is an octahedral cation and Z is a tetrahedral cation), are widely occurring rock-forming minerals. Their crystal chemistry is complex as they can accommodate a large variety of cations into their structure. Systematic structural and spectroscopic studies of synthetic micas are thus important to obtain a more complete understanding of the crystal chemical variations with changing chemical composition.
Natural trioctahedral micas have been investigated intensively by single-crystal X-ray diffraction methods and a recent review on mica crystal chemistry is found in Brigatti & Guggenheim (2002). However, studies on Fe-rich micas are rare. The Fe-bearing mica closest to the ideal composition of the mineral annite, KFe3AlSi3O10(OH)2, is that studied by Redhammer & Roth (2002), with a composition of (K0.94Na0.07)(Fe2+2.25Fe3+0.28Mn0.18Mg0.07Ti0.08) (Al0.98Fe3+0.19Si2.83)O10(OH1.85F0.15). In addition to natural trioctahedral micas, Redhammer & Roth (2002) also studied a variety of synthetic samples with various octahedral contents but containing no tetrahedral ferric iron. Other structural studies of tetraferrimicas have been carried out for CsFe3(Fe3+Si3)O10(OH)2 (Mellini et al., 1996; Comodi et al., 1999) and RbFe3(Fe3+Si3)O10(OH)2 (Comodi et al., 2003). Donnay et al. (1964) were the first workers to refine the structure of a synthetic tetraferriannite (TFA), KFe3(Fe3+Si3)O10(OH)2, from film data. However, a complete refinement was not possible and no anisotropic displacement parameters were refined, nor were the H atoms located. To date, no data are available for K-containing trioctahedral micas containing Fe exclusively in the octahedral sheet. In the course of our investigations on micas of the formula KFe3(Al1 − xFe3+xSi3)O10(OH)2, unusually large (about 500 µm long) and good-quality single crystals of the title compound were obtained. Its crystal structures at 100 K and 270 K are described here and compared with the structures of the near-end-member annite (Redhammer & Roth, 2002) and Rb- and Cs-tetraferriannite (Comodi et al., 1999, 2003).
Fig. 1 depicts part of the structure the title compound. Other polyhedral representations of the mica structure may be found in the literature (e.g. Brigatti & Guggenheim, 2002). The discussion below refers to the structure at 270 K.
The octahedral sheet of the structure contains two different M1 and M2 sites which each carry an OH group. The H atom is located 0.87 (4) Å away from atom O4 and the O—H bond is not exactly perpendicular to the (001) plane, but slightly inclined along the a axis towards the centre of the ring defined by six TO4 tetrahedra T is?. The average M1—O bond in the title compound [2.119 (1) Å; Table 2] is similar to that in the end-member tetraferriannite [2.11 (1) Å; Donnay et al., 1964). The average M1—O bond length increases with the size of the interlayer cation (Table 2) as a result of the expansion of the unit cell, not only along [001] (which is most sensitive to changes in the interlayer chemistry), but also within the (001) plane. The bond-length distortion (BLD in Table 2) of the M1 octahedron is small and similar to that found for annite and the Rb- and Cs-tetraferriannites. However, the edge-length ratio (eu/es; Toraya, 1981) and the octahedral-angle variance (OAV; Robinson et al., 1971) both indicate a significant angular distortion of the M1 site, as well as the octahedral flattening angle (ψ), which is distinctly larger than its ideal value of 54.73°. Replacing K in the interlayer by Rb or Cs increases the octahedral flattening and reduces the thickness of the octahedral sheet (Table 2). The polyhedral distortion of the M2 site is similar to that of M1. This is a general feature of all trioctahedral micas, except those containing octahedrally coordinated Al3+ (Redhammer & Roth, 2002). As for M1, the average M2—O bond length is somewhat larger than in synthetic tetraferriannite (Donnay et al., 1964).
The average T—O bond length in the title compound [1.676 (1) Å] is somewhat shorter than in the Rb- or Cs-tetraferriannites, or tetraferriphlogopite [1.680 (2) Å; Brigatti et al., 1996). This is related to the presence of tetrahedral Al3+ in addition to Si4+ and Fe3+. It has been noted by Brigatti et al. (1996) that the substitution of tetrahedral Al3+ by larger cations, such as Fe3+, produces more regular tetrahedra. This effect is also observed here, as shown by the smaller BLD and TAV Please define parameters (Table 2). As a result of the Fe3+/Al3+ tetrahedral substitution, the dimensional misfit between the octahedral and tetrahedral sheets increases but is compensated for by an increase of the tetrahedral ring-distortion angle (α angle in Table 2). This distortion of the tetrahedral sheet directly influences the coordination geometry of the interlayer cation. Whereas the inner and outer K—O bond lengths are similar when the ditrigonal distortion is small (e.g. near-end-member annites, Rb-TFA and Cs-TFA), the difference between the K—O distances increases with increasing tetrahedral rotation, α (Table 2). Due to the expansion of the octahedral sheet in the (001) plane of the Rb- and Cs-tetraferriannites, the tetrahedral ring has a nearly ideal hexagonal geometry in these structures.
At room temperature, the lattice parameters of the title compound are larger than those of pure annite (e.g. Redhammer et al., 1993; Redahmmer & Roth, 2002) but slightly smaller than those of pure tetraferriannite [a = 5.4354 (6) Å, b = 9.4186 (7) Å, c = 10.3449 (5) Å and β = 100.121 (14)°; Redhammer et al., unpublished]. These differences are attributed to the different composition of the tetrahedral sheet. Upon cooling from 270 K to 100 K, the lattice parameters decrease significantly and yield linear thermal expansion coefficients equal to 11.1 × 10−6 K−1, 10.8 × 10−6 K−1 and 21.6 × 10−6 K−1 for the a, b and c axes, respectively. These coefficients are similar to those in near-end-member phlogopite, viz. 8.6, 7.5 and 18.1 (all × 10−6 K−1; Russell & Guggenheim, 1999). The thermal expansion along c is twice as large as that within the (001) plane. The [001] direction corresponds to the stacking of the negatively charged 2:1 layers and is the direction most affected by the thermal motion of the interlayer K+ cation.
In addition to the shortening of the unit-cell parameters, the most pronounced changes upon cooling from 270 K to 100 K are observed for the interlayer region of the structure. The average K—Oinner bond shortens while the average K—Oouter bond lengthens, corresponding to an increase of the ditrigonal distortion (Table 2). This increase, as measured by a larger rotation angle α, is related to the different thermal expansion of the octahedral and tetrahedral sheets, whereby the average M—O bond lengths tend to decrease at low temperature while the average T—O bond lengths tend to increase (Table 2). Although the changes in bond length (0.003 Å) are only slightly larger than the standard uncertainty (0.001 Å), an equivalent trend was observed for a near-end-member phlogopite, KMg3AlSi3O10(OH)2, in which the average T—O bond length decreased by 0.004 Å upon heating from 293 K to 873 K (Russell & Guggenheim, 1999). The decrease in the tetrahedral angle τ upon cooling (Table 2) indicates that the tetrahedra are slightly elongated along the c axis at high temperature and compressed at low temperature.
The anisotropic atomic displacement parameters (ADP) are also affected by cooling. Atoms O1 and O2, which are shared between tetrahedra, show larger ADPs than atoms O3 and O4, which bridge tetrahedra and octahedra (Fig. 1). This suggests that the latter are more rigidly bonded than the former. The interlayer K+ cation shows a more anisotropic displacement than the octahedral cations, while the displacement of the tetrahedral cation is nearly isotropic. This behaviour is generally observed in all trioctahedral micas. Upon cooling to 100 K, the displacement decreases by ≈ 47% for the K site, ≈ 38% for the M sites, 26% for the T site and 30% for the O sites. Only minor changes are observed in the ADP max/min ratios, indicating that there is no significant change in the anisotropic behaviour of the atoms.