Buy article online - an online subscription or single-article purchase is required to access this article.
A new microporous zirconogermanate, diammonium zirconium trigermanate, (NH
4)
2ZrGe
3O
9 (FDZG-2), analogous to wadeite (K
2ZrSi
3O
9), was hydrothermally synthesized using ZrO(NO
3)
2·2H
2O as the source of zirconium and 1,4-diaminobutane as a structure-directing agent. Single-crystal X-ray diffraction analysis reveals that the framework structure is built up of cyclic trigermanate units crosslinked by ZrO
6 octahedra. The Zr atom lies at a site with
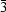
symmetry and the unique N atom of the ammonium ion lies at a site with threefold symmetry. Large cages are observed, with two NH
4+ cations in each. The structure contains intersecting six- and three-membered ring (6MR and 3MR) channels, but only the 6MR channels can accommodate the NH
4+ ions.
Supporting information
In a typical procedure, ZrO(NO3)2·2H2O (0.26 g, 0.97 mmol) was dissolved in H2O (1.07 g, 59.4 mmol), to which glycol (0.87 g, 14 mmol), GeO2 (0.24 g, 2.29 mmol) and 1,4-diaminobutane (0.50 g, 5.68 mmol) were added successively under vigorous stirring. Then a drop of 40% HF (0.05 g, 1 mmol) was added to the mixture. After stirring at room temperature for 10 h, the solution was heated at 433 K for 14 d in a Teflon-lined vessel. After the mixture was cooled to room temperature, colorless crystals were recovered. The ammonium cations in the compound are derived from the decomposition of 1,4-diaminobutane.
The locations of the highest peak and deepest hole in the difference Fourier map are 0.98 Å from Ge and 1.03 Å from Ge, respectively. H-atom positions were not included in the refinement.
Data collection: SMART (Bruker, 2001); cell refinement: SAINT (Bruker, 2001); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 1990); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: DS Viewerpro (Accelrys, 2002); software used to prepare material for publication: SHELXL97.
diammonium zirconium getmanate
top
Crystal data top
| (NH4)2ZrGe3O9 | Dx = 3.455 Mg m−3 |
| Mr = 481.01 | Mo Kα radiation, λ = 0.71073 Å |
| Hexagonal, P63/m | Cell parameters from 568 reflections |
| a = 7.117 (5) Å | θ = 5.6–23.0° |
| c = 10.542 (9) Å | µ = 10.79 mm−1 |
| V = 462.4 (6) Å3 | T = 298 K |
| Z = 2 | Prism, colorless |
| F(000) = 444 | 0.04 × 0.04 × 0.04 mm |
Data collection top
Nonius KappaCCDr
diffractometer | 367 independent reflections |
| Radiation source: fine-focus sealed tube | 280 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.085 |
| ω scans | θmax = 27.1°, θmin = 3.3° |
Absorption correction: multi-scan
(SADABS; Bruker, 2001) | h = −8→9 |
| Tmin = 0.672, Tmax = 0.672 | k = −8→9 |
| 2333 measured reflections | l = −13→11 |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.052 | H-atom parameters not refined |
| wR(F2) = 0.110 | w = 1/[σ2(Fo2) + (0.0415P)2]
where P = (Fo2 + 2Fc2)/3 |
| S = 1.13 | (Δ/σ)max < 0.001 |
| 367 reflections | Δρmax = 1.46 e Å−3 |
| 28 parameters | Δρmin = −0.84 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.009 (2) |
Crystal data top
| (NH4)2ZrGe3O9 | Z = 2 |
| Mr = 481.01 | Mo Kα radiation |
| Hexagonal, P63/m | µ = 10.79 mm−1 |
| a = 7.117 (5) Å | T = 298 K |
| c = 10.542 (9) Å | 0.04 × 0.04 × 0.04 mm |
| V = 462.4 (6) Å3 | |
Data collection top
Nonius KappaCCDr
diffractometer | 367 independent reflections |
Absorption correction: multi-scan
(SADABS; Bruker, 2001) | 280 reflections with I > 2σ(I) |
| Tmin = 0.672, Tmax = 0.672 | Rint = 0.085 |
| 2333 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.052 | 0 restraints |
| wR(F2) = 0.110 | H-atom parameters not refined |
| S = 1.13 | Δρmax = 1.46 e Å−3 |
| 367 reflections | Δρmin = −0.84 e Å−3 |
| 28 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Zr | 1.0000 | 1.0000 | 0.0000 | 0.0112 (6) | |
| Ge | 0.8761 (2) | 0.6230 (2) | 0.2500 | 0.0135 (5) | |
| O1 | 0.9877 (12) | 0.7564 (12) | 0.1128 (6) | 0.0236 (17) | |
| O2 | 0.9235 (16) | 0.4027 (16) | 0.2500 | 0.025 (3) | |
| N | 0.3333 | 0.6667 | 0.0559 (14) | 0.025 (4) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Zr | 0.0120 (8) | 0.0120 (8) | 0.0096 (12) | 0.0060 (4) | 0.000 | 0.000 |
| Ge | 0.0118 (8) | 0.0101 (8) | 0.0182 (9) | 0.0052 (7) | 0.000 | 0.000 |
| O1 | 0.029 (5) | 0.027 (4) | 0.024 (4) | 0.020 (4) | 0.004 (3) | 0.014 (3) |
| O2 | 0.014 (5) | 0.013 (5) | 0.053 (7) | 0.011 (5) | 0.000 | 0.000 |
| N | 0.025 (5) | 0.025 (5) | 0.026 (9) | 0.012 (3) | 0.000 | 0.000 |
Geometric parameters (Å, º) top
| Zr—O1i | 2.068 (7) | Ge—O1vi | 1.694 (7) |
| Zr—O1ii | 2.068 (7) | Ge—O1 | 1.694 (7) |
| Zr—O1iii | 2.068 (7) | Ge—O2vii | 1.738 (10) |
| Zr—O1 | 2.068 (7) | Ge—O2 | 1.761 (9) |
| Zr—O1iv | 2.068 (7) | O2—Geviii | 1.738 (10) |
| Zr—O1v | 2.068 (7) | | |
| | | |
| O1i—Zr—O1ii | 180.0 | O1iii—Zr—O1v | 180.0 |
| O1i—Zr—O1iii | 90.2 (3) | O1—Zr—O1v | 90.2 (3) |
| O1ii—Zr—O1iii | 89.8 (3) | O1iv—Zr—O1v | 89.8 (3) |
| O1i—Zr—O1 | 89.8 (3) | O1vi—Ge—O1 | 117.2 (5) |
| O1ii—Zr—O1 | 90.2 (3) | O1vi—Ge—O2vii | 110.6 (3) |
| O1iii—Zr—O1 | 89.8 (3) | O1—Ge—O2vii | 110.6 (3) |
| O1i—Zr—O1iv | 90.2 (3) | O1vi—Ge—O2 | 104.7 (3) |
| O1ii—Zr—O1iv | 89.8 (3) | O1—Ge—O2 | 104.7 (3) |
| O1iii—Zr—O1iv | 90.2 (3) | O2vii—Ge—O2 | 108.3 (6) |
| O1—Zr—O1iv | 180.0 | Ge—O1—Zr | 140.5 (4) |
| O1i—Zr—O1v | 89.8 (3) | Geviii—O2—Ge | 131.7 (6) |
| O1ii—Zr—O1v | 90.2 (3) | | |
| Symmetry codes: (i) y, −x+y+1, −z; (ii) −y+2, x−y+1, z; (iii) x−y+1, x, −z; (iv) −x+2, −y+2, −z; (v) −x+y+1, −x+2, z; (vi) x, y, −z+1/2; (vii) −y+1, x−y, z; (viii) −x+y+1, −x+1, z. |
Experimental details
| Crystal data |
| Chemical formula | (NH4)2ZrGe3O9 |
| Mr | 481.01 |
| Crystal system, space group | Hexagonal, P63/m |
| Temperature (K) | 298 |
| a, c (Å) | 7.117 (5), 10.542 (9) |
| V (Å3) | 462.4 (6) |
| Z | 2 |
| Radiation type | Mo Kα |
| µ (mm−1) | 10.79 |
| Crystal size (mm) | 0.04 × 0.04 × 0.04 |
| |
| Data collection |
| Diffractometer | Nonius KappaCCDr
diffractometer |
| Absorption correction | Multi-scan
(SADABS; Bruker, 2001) |
| Tmin, Tmax | 0.672, 0.672 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 2333, 367, 280 |
| Rint | 0.085 |
| (sin θ/λ)max (Å−1) | 0.641 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.052, 0.110, 1.13 |
| No. of reflections | 367 |
| No. of parameters | 28 |
| H-atom treatment | H-atom parameters not refined |
| Δρmax, Δρmin (e Å−3) | 1.46, −0.84 |
Selected geometric parameters (Å, º) top| Zr—O1 | 2.068 (7) | Ge—O2i | 1.738 (10) |
| Ge—O1 | 1.694 (7) | Ge—O2 | 1.761 (9) |
| | | |
| O1—Zr—O1ii | 90.2 (3) | O2i—Ge—O2 | 108.3 (6) |
| O1iii—Ge—O1 | 117.2 (5) | Ge—O1—Zr | 140.5 (4) |
| O1—Ge—O2i | 110.6 (3) | Geiv—O2—Ge | 131.7 (6) |
| O1—Ge—O2 | 104.7 (3) | | |
| Symmetry codes: (i) −y+1, x−y, z; (ii) −x+y+1, −x+2, z; (iii) x, y, −z+1/2; (iv) −x+y+1, −x+1, z. |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
support@iucr.org for assistance.


 journal menu
journal menu inorganic compounds
inorganic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2003/05/00/bc1005/bc1005fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2003/05/00/bc1005/bc1005fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2003/05/00/bc1005/bc1005fig3thm.gif)

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry



Ion-exchangeable materials, such as silicates and germanates, have potential applications in nuclear waste remediation (Clearfield, 1995). Recent work of our group has been directed towards the syntheses of zirconogermanates (Liu et al., 2003), of which a very limited number have been reported to date (Choisnet et al., 1973; Nosyrev et al., 1975; Ilyushin et al., 1983; Ilyushin, 1989; Pertierra et al., 1999; Li et al., 2000), although the zirconosilicate analogues have been given considerable attention during past decades (Maurice, 1949; Baussy et al., 1974; Ghose et al., 1980; Bortun et al., 1997; Lin et al., 1999; Rocha & Anderson, 2000). Wadeite (K2ZrSi3O9, P63/m, a = b = 6.893 Å, c = 10.172 Å, V = 418.6 Å3, Z = 2) is a natural zirconosilicate whose analogues, ABM3O9 (A = K, Rb; B = Ti, Sn; M = Si, Ge), have been prepared in the 1103–1273 K temperature range (Choisnet et al., 1973). We report here the synthesis and structure of its zirconogermanate analogue, (NH4)2ZrGe3O9 (denoted FDZG-2).
A single-crystal X-ray analysis reveals that the (NH4)2ZrGe3O9 structure is similar to wadeite, with Si replaced by Ge and K+ replaced by NH4+. The Ge—O bond lengths (average 1.722 Å) are close to the value expected for a Ge—O single bond (1.748 Å; Brese & O' Keeffe, 1991). The bond-valence sums (Brese & O'Keeffe, 1991) at Ge and Zr are 4.30 and 4.11, close to the expected values (4.0 and 4.0, respectively). The bond angles at the O atoms are Ge—O—Ge = 131.7 (6)° and Ge—O—Zr = 140.5 (4)°, which are smaller than the corresponding angles in wadeite.
The structure of (NH4)2ZrGe3O9 is based on an hexagonal three-dimensional framework of composition ZrGe3O9, in which Ge is tetrahedrally coordinated and Zr is octahedrally coordinated. In the structure, three GeO4 tetrahedra form cyclic trigermanate units (Ge3O9) in the ab plane, which are linked by ZrO6 octahedra (Fig. 1). The center of each cyclic trigermanate unit is located at z = 1/4, 3/4, and the Zr atoms are located on the c axis at z = 0, 1/2. This arrangement results in a cage-like topology with two kinds of cages, viz. small cages, 43, with composition Zr2Ge3O6 and large cages, 324366, with composition Zr6Ge12O15 (Fig. 2); the cage sizes are 2.38 × 2.38 × 5.29 Å3 and 4.12 × 4.12 × 10.58 Å3, respectively. In the large cages, two NH4+ ions are located near the center of a Zr triangle in the ab plane, with a slight shift of 0.58 Å above and below the Zr triangle (Fig. 2 b). The NH4+ ions not only balance the negative charges of the framework but also form weak hydrogen bonds with neighboring O atoms, which can be inferred from the three short N—O distances (2.90 Å). Because NH4+ ions were not present in the initial mixture, they may be derived from the decomposition of 1,4-diaminebutane under hydothermal conditions. The channel system of (NH4)2ZrGe3O9 is three-dimensional, with intersecting 6MR channels along the a and b axes (Fig. 3) and 3MR channels along the c axis (Fig. 1). In fact, the 6MR and 3MR channels are windows of the large cages, the free-pore diameters of which are 3.07 and 1.60 Å respectively. Because the size of the NH4+ ion is about 1.61 Å (Shannon, 1976), the movement of these ions is only free through the 6MR channels. Thus, the intersecting 6MR channels along a and b axis form two-dimensional channels for the NH4+ ions.