

 journal menu
journal menu research papers
research papers









The crystal structure of the title compound was solved from laboratory powder diffraction data in the triclinic group 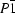 by simulated annealing using the program DASH. Since Rietveld refinements yielded inaccurate geometries the structure was finally refined by geometry optimization using energy minimization in the solid state with the DFT/plane-waves approach. The molecule is essentially planar and its Meldrum's acid moiety (2,2-dimethyl-1,3-dioxane-4,6-dione) has a flattened boat conformation. The bond orders in the molecule estimated using a natural bond-orbitals formalism correlate with the optimized bond lengths. The structure in the solid state is based on dimer units in which the molecules are held by N—H
by simulated annealing using the program DASH. Since Rietveld refinements yielded inaccurate geometries the structure was finally refined by geometry optimization using energy minimization in the solid state with the DFT/plane-waves approach. The molecule is essentially planar and its Meldrum's acid moiety (2,2-dimethyl-1,3-dioxane-4,6-dione) has a flattened boat conformation. The bond orders in the molecule estimated using a natural bond-orbitals formalism correlate with the optimized bond lengths. The structure in the solid state is based on dimer units in which the molecules are held by N—H O and C—HO hydrogen bonds in addition to electrostatic interactions. These units interact through weak C—HO hydrogen bonds. It is suggested that structure refinement by energy minimization at the DFT level of theory may in many cases successfully replace Rietveld refinement.
O and C—HO hydrogen bonds in addition to electrostatic interactions. These units interact through weak C—HO hydrogen bonds. It is suggested that structure refinement by energy minimization at the DFT level of theory may in many cases successfully replace Rietveld refinement.
O and C—HO hydrogen bonds in addition to electrostatic interactions. These units interact through weak C—HO hydrogen bonds. It is suggested that structure refinement by energy minimization at the DFT level of theory may in many cases successfully replace Rietveld refinement.Supporting information
 | Crystallographic Information File (CIF) https://doi.org/10.1107/S0108768107006568/av5081sup1.cif |
 | Rietveld powder data file (CIF format) https://doi.org/10.1107/S0108768107006568/av5081sup2.rtv |
CCDC reference: 650657
Computing details top
![[Figure 1]](http://journals.iucr.org/b/issues/2007/03/00/av5081/av5081fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/b/issues/2007/03/00/av5081/av5081fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/b/issues/2007/03/00/av5081/av5081fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/b/issues/2007/03/00/av5081/av5081fig4thm.gif)
![[Figure 5]](http://journals.iucr.org/b/issues/2007/03/00/av5081/av5081fig5thm.gif)
5-anilinomethylene-2,2-dimethyl-1,3-dioxane-4,6-dione top
Crystal data top
| C13H13NO4 | V = 621.43 Å3 |
| Mr = 247.24 | Z = 2 |
| Triclinic, P1 | The cell parameters are from structure solution by DASH. T |
| Hall symbol: -P 1 | Melting point: 156C K |
| a = 10.60310 Å | ? radiation, λ = 1.78892 Å |
| b = 11.59680 Å | θ = 3–80° |
| c = 5.50320 Å | T = 300 K |
| α = 97.8790° | , yellow |
| β = 103.8910° | × × mm |
| γ = 71.4570° |
Data collection top
| Stoe Stadi-P diffractometer | h = ?→? |
| Radiation source: fine-focus sealed tube | k = ?→? |
| Ge(111) monochromator | l = ?→? |
Crystal data top
| C13H13NO4 | β = 103.8910° |
| Mr = 247.24 | γ = 71.4570° |
| Triclinic, P1 | V = 621.43 Å3 |
| a = 10.60310 Å | Z = 2 |
| b = 11.59680 Å | ? radiation, λ = 1.78892 Å |
| c = 5.50320 Å | T = 300 K |
| α = 97.8790° | × × mm |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top
| x | y | z | Uiso*/Ueq | ||
| o1 | −0.18118 | 0.25399 | 0.11553 | 0.0500* | |
| o3 | −0.08713 | 0.34986 | −0.12582 | 0.0500* | |
| o4 | 0.12844 | 0.27052 | −0.18054 | 0.0500* | |
| o6 | −0.05771 | 0.07945 | 0.29910 | 0.0500* | |
| n1 | 0.21464 | −0.00098 | 0.30875 | 0.0500* | |
| c10 | 0.34367 | −0.08712 | 0.37325 | 0.0500* | |
| c11 | 0.44853 | −0.10153 | 0.24797 | 0.0500* | |
| c12 | 0.57416 | −0.18704 | 0.32315 | 0.0500* | |
| c13 | 0.59601 | −0.25921 | 0.52160 | 0.0500* | |
| c14 | 0.49046 | −0.24573 | 0.64318 | 0.0500* | |
| c15 | 0.36453 | −0.16019 | 0.56993 | 0.0500* | |
| c9 | 0.18392 | 0.09045 | 0.16347 | 0.0500* | |
| c5 | 0.05623 | 0.17490 | 0.10000 | 0.0500* | |
| c4 | 0.04008 | 0.26530 | −0.07128 | 0.0500* | |
| c6 | −0.05895 | 0.16536 | 0.18156 | 0.0500* | |
| c2 | −0.17708 | 0.37087 | 0.05006 | 0.0500* | |
| c7 | −0.12772 | 0.44246 | 0.28472 | 0.0500* | |
| c8 | −0.31854 | 0.43254 | −0.09035 | 0.0500* | |
| h1 | 0.13591 | −0.00757 | 0.38154 | 0.0600* | |
| h11 | 0.43256 | −0.04794 | 0.08957 | 0.0600* | |
| h12 | 0.65688 | −0.19845 | 0.22797 | 0.0600* | |
| h13 | 0.69517 | −0.32536 | 0.57951 | 0.0600* | |
| h14 | 0.50525 | −0.30095 | 0.79856 | 0.0600* | |
| h15 | 0.28141 | −0.14945 | 0.66329 | 0.0600* | |
| h9 | 0.26529 | 0.10139 | 0.08850 | 0.0600* | |
| h71 | −0.02579 | 0.39201 | 0.38223 | 0.0750* | |
| h72 | −0.12580 | 0.53058 | 0.23641 | 0.0750* | |
| h73 | −0.19648 | 0.45794 | 0.41412 | 0.0750* | |
| h81 | −0.32100 | 0.51943 | −0.15133 | 0.0750* | |
| h82 | −0.38963 | 0.45051 | 0.03538 | 0.0750* | |
| h83 | −0.34917 | 0.37437 | −0.25374 | 0.0750* |
Experimental details
| Crystal data | |
| Chemical formula | C13H13NO4 |
| Mr | 247.24 |
| Crystal system, space group | Triclinic, P1 |
| Temperature (K) | 300 |
| a, b, c (Å) | 10.60310, 11.59680, 5.50320 |
| α, β, γ (°) | 97.8790, 103.8910, 71.4570 |
| V (Å3) | 621.43 |
| Z | 2 |
| Radiation type | ?, λ = 1.78892 Å |
| µ (mm−1) | ? |
| Crystal size (mm) | × × |
| Data collection | |
| Diffractometer | Stoe Stadi-P diffractometer |
| Absorption correction | ? |
| No. of measured, independent and observed (?) reflections | ?, ?, ? |
| Rint | ? |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | ?, ?, ? |
| No. of reflections | ? |
| No. of parameters | ? |
| No. of restraints | ? |
| Δρmax, Δρmin (e Å−3) | ?, ? |



