

 journal menu
journal menu research papers
research papers









Constrained Hartree–Fock calculations have been performed to obtain wavefunctions that reproduce experimental X-ray structure-factor magnitudes for crystalline NH3 to within the limits of experimental error. Different model densities using both a single molecule and clusters of NH3 in the calculation of X-ray structure-factor magnitudes have been examined. The effects of the crystalline lattice on the experimental wavefunction of the NH3 unit can be reproducibly recovered. The construction of structure-factor magnitudes based on normally distributed random perturbations of the experimental values has also been used to gauge the accuracy of integrated atomic properties obtained from the wavefunctions, the point at which the constraint procedure should be terminated, and the approximate error in the experimental 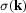 values.
values.



