Reaction of AgNO
3 and 2,2′-bipyridine (bipy) under ultrasonic treatment gave the title compound, [Ag(C
10H
8N
2)(NH
3)]NO
3. The crystal structure consists of dimers formed by two symmetry-related Ag
I–bipy monomers connected through intra-dimer π–π stacking and ligand-unsupported Ag

Ag interactions. A crystallographic
C2 axis passes through the mid-point of and is perpendicular to the Ag
Ag
i(−
x + 1,
y, −
z +
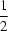
) axis. In addition, each Ag
I cation is coordinated by one chelating bipy ligand and one ammine ligand, giving a trigonal coordination environment capped by the symmetry-equivalent Ag atom. Molecules are assembled by Ag
Ag, π–π, hydrogen-bond (N—H
O and C—H
O) and weak Ag
π interactions into a three-dimensional framework. Comparing the products synthesized under different mechanical treatments, we found that reaction conditions have a significant influence on the resulting structures. The luminescence properties of the title compound are also discussed.
Supporting information
CCDC reference: 774069
All reagents and solvents were used as obtained commercially without further
purification. A mixture of AgNO3 (170 mg, 1 mmol) and 2,2'-bipyridine (156 mg, 1 mmol) was added to a methanol–water solvent mixture (15 ml, 1:2
v/v) under ultrasonic conditions, which helped to dissolve the white
precipitation. An aqueous NH3 solution (25%) was added dropwise to the
mixture to give a clear solution. The resulting solution was left to evaporate
slowly in the dark at room temperature for several weeks to give crystals of
(I) in the form of yellow prisms. The crystals were washed using deionized
water and dried in air (yield: circa 51%, based on Ag). Analysis
calculated for C20H22Ag2N8O6: C 35.01, H 3.23, N 16.33%; found: C
34.95, H. 3.14, N 16.27%.
The aromatic H atoms were generated geometrically (C—H 0.93Å) and were
allowed to ride on their parent atoms in the riding model approximations, with
Uiso(H) = 1.2Ueq(C). The positions of the ammonia H atoms
were located from difference maps and refned with the N—H distances
restrained to 0.89 (2) Å with Uiso(H) = 1.5Ueq(N).
Data collection: CrysAlis CCD (Oxford Diffraction, 2008); cell refinement: CrysAlis RED (Oxford Diffraction, 2008); data reduction: CrysAlis RED (Oxford Diffraction, 2008); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008); molecular graphics: DIAMOND (Brandenburg, 2008) and SHELXTL (Sheldrick, 2008); software used to prepare material for publication: SHELXL97 (Sheldrick, 2008), publCIF (Westrip, 2010).
Ammine(2,2'-bipyridine-
κ2N,
N')silver(I) nitrate
top
Crystal data top
| [Ag(C10H8N2)(NH3)]NO3 | F(000) = 1360 |
| Mr = 343.10 | Dx = 1.897 Mg m−3 |
| Monoclinic, C2/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -C 2yc | Cell parameters from 3049 reflections |
| a = 17.685 (9) Å | θ = 5.4–56.9° |
| b = 10.690 (5) Å | µ = 1.68 mm−1 |
| c = 12.748 (7) Å | T = 298 K |
| β = 94.457 (12)° | Prism, yellow |
| V = 2403 (2) Å3 | 0.10 × 0.08 × 0.08 mm |
| Z = 8 | |
Data collection top
Oxford Diffraction Gemini S Ultra
diffractometer | 2320 independent reflections |
| Radiation source: sealed tube | 2161 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.030 |
| Detector resolution: 16.1903 pixels mm-1 | θmax = 26.0°, θmin = 2.2° |
| ω scans | h = −17→21 |
Absorption correction: multi-scan
(CrysAlis RED; Oxford Diffraction, 2008) | k = −13→5 |
| Tmin = 0.850, Tmax = 0.877 | l = −15→14 |
| 4406 measured reflections | |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.044 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.112 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.21 | w = 1/[σ2(Fo2) + (0.0452P)2 + 2.9467P]
where P = (Fo2 + 2Fc2)/3 |
| 2320 reflections | (Δ/σ)max < 0.001 |
| 172 parameters | Δρmax = 0.85 e Å−3 |
| 6 restraints | Δρmin = −0.59 e Å−3 |
Crystal data top
| [Ag(C10H8N2)(NH3)]NO3 | V = 2403 (2) Å3 |
| Mr = 343.10 | Z = 8 |
| Monoclinic, C2/c | Mo Kα radiation |
| a = 17.685 (9) Å | µ = 1.68 mm−1 |
| b = 10.690 (5) Å | T = 298 K |
| c = 12.748 (7) Å | 0.10 × 0.08 × 0.08 mm |
| β = 94.457 (12)° | |
Data collection top
Oxford Diffraction Gemini S Ultra
diffractometer | 2320 independent reflections |
Absorption correction: multi-scan
(CrysAlis RED; Oxford Diffraction, 2008) | 2161 reflections with I > 2σ(I) |
| Tmin = 0.850, Tmax = 0.877 | Rint = 0.030 |
| 4406 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.044 | 6 restraints |
| wR(F2) = 0.112 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.21 | Δρmax = 0.85 e Å−3 |
| 2320 reflections | Δρmin = −0.59 e Å−3 |
| 172 parameters | |
Special details top
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell esds are taken
into account individually in the estimation of esds in distances, angles
and torsion angles; correlations between esds in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell esds is used for estimating esds involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and
goodness of fit S are based on F2, conventional R-factors R are based
on F, with F set to zero for negative F2. The threshold expression of
F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is
not relevant to the choice of reflections for refinement. R-factors based
on F2 are statistically about twice as large as those based on F, and R-
factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Ag1 | 0.47287 (2) | 0.67735 (3) | 0.13367 (3) | 0.04812 (18) | |
| C1 | 0.6485 (3) | 0.5882 (4) | 0.1571 (3) | 0.0401 (10) | |
| H1 | 0.6573 | 0.6733 | 0.1674 | 0.048* | |
| C2 | 0.7100 (3) | 0.5087 (5) | 0.1566 (4) | 0.0492 (11) | |
| H2 | 0.7591 | 0.5394 | 0.1680 | 0.059* | |
| C3 | 0.6974 (3) | 0.3850 (4) | 0.1391 (4) | 0.0456 (11) | |
| H3 | 0.7379 | 0.3297 | 0.1379 | 0.055* | |
| C4 | 0.6239 (2) | 0.3421 (4) | 0.1233 (4) | 0.0362 (9) | |
| H4 | 0.6144 | 0.2578 | 0.1099 | 0.043* | |
| C5 | 0.5642 (2) | 0.4260 (3) | 0.1275 (3) | 0.0307 (8) | |
| C6 | 0.4836 (2) | 0.3854 (3) | 0.1149 (3) | 0.0275 (8) | |
| C7 | 0.4630 (2) | 0.2606 (4) | 0.1049 (3) | 0.0362 (9) | |
| H7 | 0.5001 | 0.1988 | 0.1054 | 0.043* | |
| C8 | 0.3873 (3) | 0.2284 (4) | 0.0944 (4) | 0.0427 (10) | |
| H8 | 0.3729 | 0.1448 | 0.0884 | 0.051* | |
| C9 | 0.3337 (3) | 0.3205 (4) | 0.0928 (4) | 0.0411 (10) | |
| H9 | 0.2823 | 0.3013 | 0.0856 | 0.049* | |
| C10 | 0.3579 (2) | 0.4425 (4) | 0.1021 (3) | 0.0368 (9) | |
| H10 | 0.3214 | 0.5053 | 0.1009 | 0.044* | |
| N1 | 0.5773 (2) | 0.5483 (3) | 0.1434 (3) | 0.0329 (7) | |
| N2 | 0.43030 (19) | 0.4753 (3) | 0.1126 (3) | 0.0328 (7) | |
| N3 | 0.4346 (2) | 0.8667 (3) | 0.1352 (3) | 0.0403 (8) | |
| H3A | 0.472 (2) | 0.920 (4) | 0.135 (3) | 0.060* | |
| H3B | 0.411 (2) | 0.886 (5) | 0.192 (2) | 0.060* | |
| H3C | 0.401 (2) | 0.887 (5) | 0.082 (3) | 0.060* | |
| N4 | 0.86176 (19) | 0.4851 (3) | 0.3840 (3) | 0.0371 (8) | |
| O1 | 0.9264 (2) | 0.5243 (3) | 0.3790 (5) | 0.099 (2) | |
| O2 | 0.82189 (19) | 0.5343 (3) | 0.4472 (3) | 0.0529 (8) | |
| O3 | 0.83974 (19) | 0.3966 (3) | 0.3291 (3) | 0.0533 (8) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Ag1 | 0.0854 (3) | 0.0242 (2) | 0.0354 (2) | 0.01682 (14) | 0.00884 (19) | 0.00001 (12) |
| C1 | 0.054 (3) | 0.031 (2) | 0.036 (2) | −0.0101 (18) | 0.0120 (19) | 0.0048 (17) |
| C2 | 0.042 (3) | 0.052 (3) | 0.054 (3) | −0.010 (2) | 0.009 (2) | 0.013 (2) |
| C3 | 0.042 (2) | 0.044 (2) | 0.052 (3) | 0.0081 (19) | 0.006 (2) | 0.011 (2) |
| C4 | 0.036 (2) | 0.0322 (19) | 0.041 (2) | 0.0062 (16) | 0.0069 (18) | 0.0066 (17) |
| C5 | 0.042 (2) | 0.0244 (17) | 0.0260 (19) | 0.0018 (15) | 0.0056 (16) | 0.0037 (14) |
| C6 | 0.038 (2) | 0.0220 (17) | 0.0225 (17) | 0.0070 (14) | 0.0037 (15) | 0.0002 (14) |
| C7 | 0.042 (2) | 0.0275 (19) | 0.040 (2) | 0.0072 (16) | 0.0076 (18) | −0.0060 (17) |
| C8 | 0.049 (3) | 0.033 (2) | 0.047 (3) | −0.0042 (18) | 0.007 (2) | −0.0055 (19) |
| C9 | 0.037 (2) | 0.048 (3) | 0.038 (2) | −0.0024 (18) | 0.0035 (19) | −0.0060 (18) |
| C10 | 0.042 (2) | 0.039 (2) | 0.030 (2) | 0.0127 (18) | 0.0017 (17) | −0.0022 (17) |
| N1 | 0.047 (2) | 0.0262 (15) | 0.0269 (16) | −0.0006 (14) | 0.0083 (14) | 0.0004 (13) |
| N2 | 0.041 (2) | 0.0281 (16) | 0.0290 (17) | 0.0107 (14) | −0.0002 (14) | −0.0034 (13) |
| N3 | 0.050 (2) | 0.0258 (17) | 0.046 (2) | 0.0099 (15) | 0.0082 (17) | −0.0006 (15) |
| N4 | 0.0317 (18) | 0.0240 (15) | 0.056 (2) | 0.0007 (13) | 0.0035 (16) | 0.0020 (15) |
| O1 | 0.051 (2) | 0.039 (2) | 0.215 (6) | −0.0162 (17) | 0.057 (3) | −0.038 (3) |
| O2 | 0.0487 (19) | 0.0536 (19) | 0.057 (2) | 0.0024 (15) | 0.0091 (16) | −0.0148 (17) |
| O3 | 0.058 (2) | 0.0475 (18) | 0.055 (2) | −0.0100 (16) | 0.0092 (16) | −0.0213 (16) |
Geometric parameters (Å, º) top
| Ag1—N3 | 2.135 (4) | C6—C7 | 1.387 (6) |
| Ag1—N2 | 2.296 (3) | C7—C8 | 1.378 (6) |
| Ag1—N1 | 2.302 (3) | C7—H7 | 0.9300 |
| Ag1—Ag1i | 3.0456 (16) | C8—C9 | 1.366 (6) |
| C1—N1 | 1.328 (6) | C8—H8 | 0.9300 |
| C1—C2 | 1.380 (7) | C9—C10 | 1.374 (6) |
| C1—H1 | 0.9300 | C9—H9 | 0.9300 |
| C2—C3 | 1.357 (7) | C10—N2 | 1.325 (6) |
| C2—H2 | 0.9300 | C10—H10 | 0.9300 |
| C3—C4 | 1.378 (6) | N3—H3A | 0.872 (19) |
| C3—H3 | 0.9300 | N3—H3B | 0.891 (19) |
| C4—C5 | 1.389 (5) | N3—H3C | 0.884 (19) |
| C4—H4 | 0.9300 | N4—O3 | 1.223 (4) |
| C5—N1 | 1.340 (5) | N4—O1 | 1.223 (5) |
| C5—C6 | 1.487 (6) | N4—O2 | 1.229 (5) |
| C6—N2 | 1.345 (5) | | |
| | | |
| N3—Ag1—N2 | 142.34 (14) | C6—C7—H7 | 120.1 |
| N3—Ag1—N1 | 145.25 (14) | C9—C8—C7 | 119.2 (4) |
| N2—Ag1—N1 | 72.36 (12) | C9—C8—H8 | 120.4 |
| N3—Ag1—Ag1i | 93.86 (11) | C7—C8—H8 | 120.4 |
| N2—Ag1—Ag1i | 100.92 (8) | C8—C9—C10 | 118.2 (4) |
| N1—Ag1—Ag1i | 75.91 (8) | C8—C9—H9 | 120.9 |
| N1—C1—C2 | 122.7 (4) | C10—C9—H9 | 120.9 |
| N1—C1—H1 | 118.6 | N2—C10—C9 | 123.5 (4) |
| C2—C1—H1 | 118.6 | N2—C10—H10 | 118.2 |
| C3—C2—C1 | 118.8 (4) | C9—C10—H10 | 118.2 |
| C3—C2—H2 | 120.6 | C1—N1—C5 | 119.0 (4) |
| C1—C2—H2 | 120.6 | C1—N1—Ag1 | 124.3 (3) |
| C2—C3—C4 | 119.3 (4) | C5—N1—Ag1 | 116.6 (3) |
| C2—C3—H3 | 120.3 | C10—N2—C6 | 118.9 (3) |
| C4—C3—H3 | 120.3 | C10—N2—Ag1 | 124.5 (3) |
| C3—C4—C5 | 119.3 (4) | C6—N2—Ag1 | 116.5 (3) |
| C3—C4—H4 | 120.3 | Ag1—N3—H3A | 112 (4) |
| C5—C4—H4 | 120.3 | Ag1—N3—H3B | 113 (3) |
| N1—C5—C4 | 120.9 (4) | H3A—N3—H3B | 105 (3) |
| N1—C5—C6 | 117.0 (3) | Ag1—N3—H3C | 114 (4) |
| C4—C5—C6 | 122.1 (3) | H3A—N3—H3C | 107 (3) |
| N2—C6—C7 | 120.4 (4) | H3B—N3—H3C | 103 (3) |
| N2—C6—C5 | 117.3 (3) | O3—N4—O1 | 119.5 (4) |
| C7—C6—C5 | 122.3 (3) | O3—N4—O2 | 122.2 (4) |
| C8—C7—C6 | 119.8 (4) | O1—N4—O2 | 118.2 (4) |
| C8—C7—H7 | 120.1 | | |
| | | |
| N1—C5—C6—N2 | 4.8 (5) | N3—Ag1—Ag1i—N3i | −36.3 (2) |
| Symmetry code: (i) −x+1, y, −z+1/2. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N3—H3A···O1ii | 0.87 (2) | 2.14 (2) | 2.998 (6) | 170 (5) |
| N3—H3B···O3iii | 0.89 (2) | 2.23 (2) | 3.110 (5) | 169 (4) |
| N3—H3C···O2iv | 0.88 (2) | 2.30 (2) | 3.177 (5) | 172 (5) |
| C1—H1···O3ii | 0.93 | 2.39 | 3.307 (5) | 169 |
| C7—H7···O1v | 0.93 | 2.27 | 3.192 (5) | 169 |
| C10—H10···O2i | 0.93 | 2.58 | 3.340 (6) | 139 |
| Symmetry codes: (i) −x+1, y, −z+1/2; (ii) −x+3/2, y+1/2, −z+1/2; (iii) x−1/2, y+1/2, z; (iv) x−1/2, −y+3/2, z−1/2; (v) −x+3/2, y−1/2, −z+1/2. |
Experimental details
| Crystal data |
| Chemical formula | [Ag(C10H8N2)(NH3)]NO3 |
| Mr | 343.10 |
| Crystal system, space group | Monoclinic, C2/c |
| Temperature (K) | 298 |
| a, b, c (Å) | 17.685 (9), 10.690 (5), 12.748 (7) |
| β (°) | 94.457 (12) |
| V (Å3) | 2403 (2) |
| Z | 8 |
| Radiation type | Mo Kα |
| µ (mm−1) | 1.68 |
| Crystal size (mm) | 0.10 × 0.08 × 0.08 |
| |
| Data collection |
| Diffractometer | Oxford Diffraction Gemini S Ultra
diffractometer |
| Absorption correction | Multi-scan
(CrysAlis RED; Oxford Diffraction, 2008) |
| Tmin, Tmax | 0.850, 0.877 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 4406, 2320, 2161 |
| Rint | 0.030 |
| (sin θ/λ)max (Å−1) | 0.617 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.044, 0.112, 1.21 |
| No. of reflections | 2320 |
| No. of parameters | 172 |
| No. of restraints | 6 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.85, −0.59 |
Selected geometric parameters (Å, º) top| Ag1—N3 | 2.135 (4) | Ag1—N1 | 2.302 (3) |
| Ag1—N2 | 2.296 (3) | Ag1—Ag1i | 3.0456 (16) |
| | | |
| N3—Ag1—N2 | 142.34 (14) | N3—Ag1—Ag1i | 93.86 (11) |
| N3—Ag1—N1 | 145.25 (14) | N2—Ag1—Ag1i | 100.92 (8) |
| N2—Ag1—N1 | 72.36 (12) | N1—Ag1—Ag1i | 75.91 (8) |
| Symmetry code: (i) −x+1, y, −z+1/2. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N3—H3A···O1ii | 0.872 (19) | 2.14 (2) | 2.998 (6) | 170 (5) |
| N3—H3B···O3iii | 0.891 (19) | 2.23 (2) | 3.110 (5) | 169 (4) |
| N3—H3C···O2iv | 0.884 (19) | 2.30 (2) | 3.177 (5) | 172 (5) |
| C1—H1···O3ii | 0.93 | 2.39 | 3.307 (5) | 169.4 |
| C7—H7···O1v | 0.93 | 2.27 | 3.192 (5) | 168.8 |
| C10—H10···O2i | 0.93 | 2.58 | 3.340 (6) | 139.2 |
| Symmetry codes: (i) −x+1, y, −z+1/2; (ii) −x+3/2, y+1/2, −z+1/2; (iii) x−1/2, y+1/2, z; (iv) x−1/2, −y+3/2, z−1/2; (v) −x+3/2, y−1/2, −z+1/2. |


 journal menu
journal menu metal-organic compounds
metal-organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2010/03/00/mx3026/mx3026fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2010/03/00/mx3026/mx3026fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2010/03/00/mx3026/mx3026fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/c/issues/2010/03/00/mx3026/mx3026fig4thm.gif)




Self-assembled coordination structures are one of the most attractive areas of materials research due to their intriguing structural topologies and functional properties such as molecular adsorption, magnetism, and luminescence (Biradha et al., 2006; Wu et al., 2009; Blake, Brooks et al., 1999; Blake, Champness et al., 1999; Evans & Lin, 2002; Kitagawa et al., 2004; Yaghi et al., 2003), and much attention is focused on their design and construction. However, the factors that govern the formation of such complexes are complicated and include not only the inherent properties of metal ions and ligand structure, but also anion-directed interactions as well as reaction conditions (Bu et al., 2003; Kong et al., 2008, 2009). In addition to covalent bonds, noncovalent interactions such as Ag···Ag, π–π, hydrogen bonds and cation···π interactions also play important roles in controlling molecular packing (Pedireddi et al., 1996; Kolotuchin et al., 1995; Li et al., 2006; Sun et al., 2003; Lough et al., 2000; Massoud & Langer, 2009; Goodgame et al., 2002). Because the central C—C bond of bipy (bipyridine) can rotate freely, bipy cannot just be regarded as a chelating ligand (Kaes et al., 2000; Marchetti et al., 2007), but also as a potential spacer between metal centers by acting as a bridging ligand with an anti configuration (Yu et al., 2007; Forniés et al., 1993). Therefore, bipy and its derivatives are widely used in the construction of novel AgI-containing complexes incorporating diverse supramolecular interactions (Hung-Low et al., 2009; Ye et al., 2005). Recently, we have pursued systematic investigations into the assembly of AgI cations with different angular and linear bipodal N-donor ligands, such as aminopyrimidine and aminopyrazine (e.g. Luo, Huang, Chen et al., 2008; Luo, Huang, Zhang et al., 2008; Luo et al., 2009; Sun, Luo, Huang et al., 2009; Sun, Luo, Xu et al., 2009; Sun, Luo, Zhang et al., 2009), with the principal aim of obtaining supramolecular complexes or multifunctional coordination polymers. In an attempt to exploit the influence of synthesis conditions on the structures of the AgNO3–bipy system, we successfully obtained the title compound, (I), and the known coordination polymer, (II) {catena-[(µ2-nitrato-O,O,O')-(2,2'-bipyridine-N,N')-silver(I)]; Bowmaker et al., 2005} in the same solvent system.
The asymmetric unit of (I) contains one AgI cation, one bipy ligand, one coordinated ammonia molecule and one nitrate anion. As shown in Fig. 1, the AgI cation is coordinated in a trigonal planar fashion by three N atoms from one bipy ligand and one ammonia molecule, with bond angles ranging from 72.36 (12) to 145.23 (14) °. The Ag – Nbipy bond lengths are identical within experimental error (Table 1) and comparable to reported values (Oxtoby et al., 2002; Fan et al., 2007; Nicola et al., 2007). The pyridyl rings of the bipy are nearly coplanar with a twist angle of 4.8 (5)°. With an Ag—N bond length of 2.135 (4) Å, the coordinated ammonia molecule plays a role as terminator, obstructing aggregation of (I). Because of the labile nature of the this Ag—N bond, the presence of the ammonia in the coordination sphere of the metal center offers a potential coordination site in the molecule. Two symmetry-related AgI--bipy monomers aggregate to a dimer with a head-to-head arrangement through intra-dimer π–π stacking and a ligand-unsupported Ag···Ag interaction (Tong et al., 1999), where the Ag···Agi interaction has a distance of 3.0456 (16) Å. This is significantly shorter than twice the van der Waals radius of AgI (3.44 Å; Bondi, 1964) and the completed coordination sphere of the Ag centers can thus be described as capped trigonal planar. The other cases where such short contacts exist between non-bridged AgI cations are found in Ag(imidazole)2ClO4 (Eastland et al., 1980) and [Cu(ethylenediamine)3][Ag2(CN)4] (Kappenstein et al.,1988). A crystallographic C2 axis passes perpendicularly through the midpoint of this Ag···Agi axis. This weak bonding interaction between two d10 cations is possible via the participation of 5s and 5p orbitals which are close in energy to the 4d orbitals. Intra-dimer π–π stacking also contributes to the reinforcement of this Ag···Ag interaction (Venkatalakshmi et al., 1992). Moreover, the π–π stacking [Cg1···Cg2i = 3.628 (3) Å (intra-dimer) and Cg1···Cg2vi = 3.711 (3) Å (inter-dimer); Cg1 and Cg2 are the centroids of the N1/C1–C5 and N2/C6–10 rings, respectively; symmetry codes: (i) – x + 1, y, – z + 1/2; (vi) – x + 1 , – y + 1 , – z; Fig. 2] and weak Ag···C interactions [Ag1···C6vi = 3.386 (4) and Ag1···C7vi = 3.393 (5) Å; symmetry code: (vi) – x + 1 , – y + 1 , – z] act as a `glue' to reinforce the dimers, forming a column along the c axis, in which the dimers are arranged in a head-to-tail orientation. In addition, the nitrate anion acts as an acceptor and is hydrogen bonded to three different symmetry equivalents of the ammonia molecules (Table 2). Non-classic Cpyridyl—H···O hydrogen bonds [average C···O distance: 3.280 (5) Å; Table 2] and classic N—H···O hydrogen bonds link adjacent columns to form the resulting three-dimensional supramolecular framework (Fig. 3).
The effects of the synthesis conditions on the structure of the AgNO3–bipy system were investigated in the ultrasonic and stirred methods with the same solvent system (methanol-water; 15 ml, 1:2 v/v). Under the stirred condition, we could only obtain coordination polymer (II), first reported by the A. H. White group (Bowmaker et al., 2005). In the structure of (II), the nitrate not only acts as a coordinated anion but also as a bridging anion to link AgI–bipy cationic units into one-dimensional zigzag chains. The difference between the structures of (I) and (II) originates mainly from the different mechanical treatments which cause the ammonia molecule and nitrate anion to play different roles in the construction of (I) and (II).
It is known that the free bipy molecule displays a weak luminescence at circa 530 nm in the solid state at room temperature. As shown in Fig. 4, compound (I) exhibits an intense emission maximum at 469 nm upon excitation at 345 nm, which may be attributed to the intraligand emission from the bipy (Wang et al., 2004). Compared with that of the free bipy molecule, the blue shift and the luminescent enhancement of the emission at 469 nm may be due to the chelation of the bipy ligand to the AgI, which effectively increases the rigidity and coplanarity of the ligand and reduces the loss of energy by nonradiative decay of the intraligand emission excited state (Zhang et al., 2003; Qian & Wang 2002).