2-Amino-5-nitrothiazole crystallizes from solution in ethanol as a monosolvate, C
3H
3N
3O
2S·C
2H
6O, in which the thiazole component has a strongly polarized molecular–electronic structure. The thiazole molecules are linked into centrosymmetric dimers by paired N—H

N hydrogen bonds [H
N = 2.09 Å, N
N = 2.960 (6) Å and N—H
N = 169°], and these dimers are linked by the ethanol molecules,
via a two-centred N—H
O hydrogen bond [H
O = 1.98 Å, N
O = 2.838 (5) Å and N—H
O = 164°] and a planar asymmetric three-centred O—H
(O)
2 hydrogen bond [H
O = 2.07 and 2.53 Å, O
O = 2.900 (5) and 3.188 (5) Å, O—H
O = 169 and 136°, and O
H
O = 55°], into sheets built from alternating
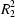
(8) and
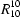
(38) rings. These sheets are triply interwoven.
Supporting information
CCDC reference: 231053
A sample of 2-amino-5-nitrothiazole was purchased from Aldrich. Crystals of the ethanol solvate, (VI), suitable for single-crystal X-ray diffraction were grown by slow evaporation of a solution in ethanol. The crystal quality was consistently poor, as shown by the high value of the merging index.
Space group P21/c was uniquely assigned from the systematic absences. All H atoms were located from difference maps and then treated as riding atoms, with C—H distances of 0.95 Å (ring CH), 0.98 Å (CH3) and 0.99 Å (CH2), N—H distances of 0.88 Å, and O—H distances of 0.84 Å.
Data collection: KappaCCD Server Software (Nonius, 1997); cell refinement: DENZO-SMN (Otwinowski & Minor, 1997); data reduction: DENZO-SMN (Otwinowski & Minor, 1997); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: PLATON (Spek, 200); software used to prepare material for publication: SHELXL97 (Sheldrick, 1997) and PRPKAPPA (Ferguson, 1999).
2-Amino-5-nitrothiazole monoethanol solvate
top
Crystal data top
| C3H3N3O2S·C2H6O | F(000) = 400 |
| Mr = 191.22 | Dx = 1.512 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 1602 reflections |
| a = 13.562 (2) Å | θ = 3.1–26.0° |
| b = 5.3311 (8) Å | µ = 0.36 mm−1 |
| c = 13.142 (2) Å | T = 120 K |
| β = 117.900 (6)° | Needle, colourless |
| V = 839.7 (2) Å3 | 0.20 × 0.05 × 0.02 mm |
| Z = 4 | |
Data collection top
Nonius KappaCCD
diffractometer | 1602 independent reflections |
| Radiation source: rotating anode | 877 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.135 |
| ϕ scans, and ω scans with κ offsets | θmax = 26.0°, θmin = 3.1° |
Absorption correction: multi-scan
DENZO-SMN (Otwinowski & Minor, 1997) | h = −16→16 |
| Tmin = 0.910, Tmax = 0.993 | k = −6→6 |
| 5482 measured reflections | l = −16→15 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.072 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.181 | H-atom parameters constrained |
| S = 0.99 | w = 1/[σ2(Fo2) + (0.0791P)2]
where P = (Fo2 + 2Fc2)/3 |
| 1602 reflections | (Δ/σ)max = 0.001 |
| 111 parameters | Δρmax = 0.95 e Å−3 |
| 0 restraints | Δρmin = −0.36 e Å−3 |
Crystal data top
| C3H3N3O2S·C2H6O | V = 839.7 (2) Å3 |
| Mr = 191.22 | Z = 4 |
| Monoclinic, P21/c | Mo Kα radiation |
| a = 13.562 (2) Å | µ = 0.36 mm−1 |
| b = 5.3311 (8) Å | T = 120 K |
| c = 13.142 (2) Å | 0.20 × 0.05 × 0.02 mm |
| β = 117.900 (6)° | |
Data collection top
Nonius KappaCCD
diffractometer | 1602 independent reflections |
Absorption correction: multi-scan
DENZO-SMN (Otwinowski & Minor, 1997) | 877 reflections with I > 2σ(I) |
| Tmin = 0.910, Tmax = 0.993 | Rint = 0.135 |
| 5482 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.072 | 0 restraints |
| wR(F2) = 0.181 | H-atom parameters constrained |
| S = 0.99 | Δρmax = 0.95 e Å−3 |
| 1602 reflections | Δρmin = −0.36 e Å−3 |
| 111 parameters | |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| C1 | 0.3344 (4) | −0.0905 (9) | 0.5364 (4) | 0.0294 (12) | |
| C2 | 0.3698 (4) | 0.2328 (9) | 0.4305 (4) | 0.0322 (12) | |
| C3 | 0.4228 (4) | 0.0601 (9) | 0.6007 (4) | 0.0315 (12) | |
| N1 | 0.2957 (3) | −0.2868 (7) | 0.5767 (3) | 0.0338 (10) | |
| N2 | 0.3705 (3) | 0.3908 (7) | 0.3542 (3) | 0.0335 (11) | |
| N3 | 0.4439 (3) | 0.2444 (7) | 0.5433 (3) | 0.0335 (11) | |
| O1 | 0.3418 (3) | −0.3402 (6) | 0.6804 (3) | 0.0412 (10) | |
| O2 | 0.2118 (3) | −0.4074 (6) | 0.5054 (3) | 0.0397 (10) | |
| S | 0.27102 (10) | −0.0067 (2) | 0.39302 (10) | 0.0335 (4) | |
| C4 | 0.0716 (4) | 0.2864 (10) | 0.1230 (4) | 0.0435 (14) | |
| C5 | 0.0552 (5) | 0.4958 (10) | 0.1903 (4) | 0.0488 (15) | |
| O3 | 0.1821 (3) | 0.3113 (7) | 0.1364 (3) | 0.0490 (11) | |
| H3 | 0.4670 | 0.0369 | 0.6813 | 0.038* | |
| H2A | 0.4208 | 0.5107 | 0.3760 | 0.040* | |
| H2B | 0.3205 | 0.3767 | 0.2814 | 0.040* | |
| H4A | 0.0627 | 0.1219 | 0.1527 | 0.052* | |
| H4B | 0.0159 | 0.2987 | 0.0407 | 0.052* | |
| H5A | 0.1068 | 0.4739 | 0.2723 | 0.073* | |
| H5B | −0.0218 | 0.4929 | 0.1783 | 0.073* | |
| H5C | 0.0699 | 0.6569 | 0.1640 | 0.073* | |
| H3A | 0.1972 | 0.1859 | 0.1075 | 0.059* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| C1 | 0.031 (3) | 0.030 (2) | 0.029 (3) | 0.000 (2) | 0.016 (2) | 0.002 (2) |
| C2 | 0.026 (3) | 0.033 (3) | 0.036 (3) | 0.003 (2) | 0.013 (2) | −0.001 (2) |
| C3 | 0.025 (3) | 0.040 (3) | 0.026 (3) | 0.005 (2) | 0.010 (2) | 0.000 (2) |
| N1 | 0.030 (2) | 0.043 (2) | 0.030 (2) | −0.001 (2) | 0.016 (2) | 0.003 (2) |
| N2 | 0.031 (2) | 0.036 (2) | 0.032 (2) | −0.0023 (19) | 0.0130 (19) | 0.0051 (19) |
| N3 | 0.031 (2) | 0.028 (2) | 0.038 (3) | −0.0002 (19) | 0.014 (2) | 0.0012 (19) |
| O1 | 0.045 (2) | 0.045 (2) | 0.029 (2) | −0.0037 (18) | 0.0136 (17) | 0.0056 (16) |
| O2 | 0.031 (2) | 0.043 (2) | 0.036 (2) | −0.0130 (17) | 0.0078 (17) | −0.0016 (16) |
| S | 0.0304 (7) | 0.0343 (7) | 0.0299 (7) | −0.0042 (6) | 0.0092 (5) | 0.0016 (6) |
| C4 | 0.037 (3) | 0.051 (3) | 0.039 (3) | 0.002 (3) | 0.015 (3) | 0.004 (3) |
| C5 | 0.040 (3) | 0.062 (4) | 0.048 (3) | 0.005 (3) | 0.023 (3) | −0.009 (3) |
| O3 | 0.037 (2) | 0.053 (3) | 0.059 (3) | −0.0042 (18) | 0.0242 (19) | −0.0219 (19) |
Geometric parameters (Å, º) top
| S—C1 | 1.724 (5) | N2—H2A | 0.88 |
| C1—C3 | 1.359 (7) | N2—H2B | 0.88 |
| C3—N3 | 1.349 (6) | C4—O3 | 1.432 (6) |
| N3—C2 | 1.348 (6) | C4—C5 | 1.503 (7) |
| S—C2 | 1.746 (5) | C4—H4A | 0.99 |
| C1—N1 | 1.382 (6) | C4—H4B | 0.99 |
| C2—N2 | 1.312 (6) | C5—H5A | 0.98 |
| C3—H3 | 0.95 | C5—H5B | 0.98 |
| N1—O1 | 1.238 (5) | C5—H5C | 0.98 |
| N1—O2 | 1.260 (5) | O3—H3A | 0.84 |
| | | |
| C1—S—C2 | 87.5 (2) | C2—N2—H2B | 120.0 |
| S—C2—N3 | 115.0 (4) | H2A—N2—H2B | 120.0 |
| C2—N3—C3 | 109.7 (4) | O3—C4—C5 | 107.1 (4) |
| N3—C3—C1 | 116.0 (4) | O3—C4—H4A | 110.3 |
| C3—C1—S | 111.7 (4) | C5—C4—H4A | 110.3 |
| C3—C1—N1 | 126.2 (4) | O3—C4—H4B | 110.3 |
| N1—C1—S | 122.0 (3) | C5—C4—H4B | 110.3 |
| N2—C2—N3 | 122.8 (4) | H4A—C4—H4B | 108.6 |
| N2—C2—S | 122.2 (4) | C4—C5—H5A | 109.5 |
| N3—C3—H3 | 122.0 | C4—C5—H5B | 109.5 |
| C1—C3—H3 | 122.0 | H5A—C5—H5B | 109.5 |
| O1—N1—O2 | 121.1 (4) | C4—C5—H5C | 109.5 |
| O1—N1—C1 | 120.6 (4) | H5A—C5—H5C | 109.5 |
| O2—N1—C1 | 118.3 (4) | H5B—C5—H5C | 109.5 |
| C2—N2—H2A | 120.0 | C4—O3—H3A | 109.5 |
| | | |
| N1—C1—C3—N3 | −178.5 (5) | S—C2—N3—C3 | 0.4 (5) |
| S—C1—C3—N3 | 0.7 (6) | C1—C3—N3—C2 | −0.7 (6) |
| C3—C1—N1—O1 | 0.3 (8) | C3—C1—S—C2 | −0.4 (4) |
| S—C1—N1—O1 | −178.8 (3) | N1—C1—S—C2 | 178.9 (5) |
| C3—C1—N1—O2 | 179.5 (5) | N2—C2—S—C1 | −179.3 (4) |
| S—C1—N1—O2 | 0.4 (6) | N3—C2—S—C1 | 0.0 (4) |
| N2—C2—N3—C3 | 179.7 (5) | | |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H2A···N3i | 0.88 | 2.09 | 2.960 (6) | 169 |
| N2—H2B···O3 | 0.88 | 1.98 | 2.838 (5) | 164 |
| O3—H3A···O1ii | 0.84 | 2.53 | 3.188 (5) | 136 |
| O3—H3A···O2ii | 0.84 | 2.07 | 2.900 (5) | 169 |
| C3—H3···O1iii | 0.95 | 2.46 | 3.187 (6) | 133 |
| Symmetry codes: (i) −x+1, −y+1, −z+1; (ii) x, −y−1/2, z−1/2; (iii) −x+1, y+1/2, −z+3/2. |
Experimental details
| Crystal data |
| Chemical formula | C3H3N3O2S·C2H6O |
| Mr | 191.22 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 120 |
| a, b, c (Å) | 13.562 (2), 5.3311 (8), 13.142 (2) |
| β (°) | 117.900 (6) |
| V (Å3) | 839.7 (2) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.36 |
| Crystal size (mm) | 0.20 × 0.05 × 0.02 |
| |
| Data collection |
| Diffractometer | Nonius KappaCCD
diffractometer |
| Absorption correction | Multi-scan
DENZO-SMN (Otwinowski & Minor, 1997) |
| Tmin, Tmax | 0.910, 0.993 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 5482, 1602, 877 |
| Rint | 0.135 |
| (sin θ/λ)max (Å−1) | 0.617 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.072, 0.181, 0.99 |
| No. of reflections | 1602 |
| No. of parameters | 111 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.95, −0.36 |
Selected geometric parameters (Å, º) top| S—C1 | 1.724 (5) | S—C2 | 1.746 (5) |
| C1—C3 | 1.359 (7) | C1—N1 | 1.382 (6) |
| C3—N3 | 1.349 (6) | C2—N2 | 1.312 (6) |
| N3—C2 | 1.348 (6) | | |
| | | |
| C1—S—C2 | 87.5 (2) | N3—C3—C1 | 116.0 (4) |
| S—C2—N3 | 115.0 (4) | C3—C1—S | 111.7 (4) |
| C2—N3—C3 | 109.7 (4) | | |
| | | |
| S—C1—N1—O1 | −178.8 (3) | S—C1—N1—O2 | 0.4 (6) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H2A···N3i | 0.88 | 2.09 | 2.960 (6) | 169 |
| N2—H2B···O3 | 0.88 | 1.98 | 2.838 (5) | 164 |
| O3—H3A···O1ii | 0.84 | 2.53 | 3.188 (5) | 136 |
| O3—H3A···O2ii | 0.84 | 2.07 | 2.900 (5) | 169 |
| C3—H3···O1iii | 0.95 | 2.46 | 3.187 (6) | 133 |
| Symmetry codes: (i) −x+1, −y+1, −z+1; (ii) x, −y−1/2, z−1/2; (iii) −x+1, y+1/2, −z+3/2. |


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2004/01/00/gg1195/gg1195fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2004/01/00/gg1195/gg1195fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2004/01/00/gg1195/gg1195fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/c/issues/2004/01/00/gg1195/gg1195fig4thm.gif)




Supramolecular aggregation in simple nitroanilines is dominated by N—H···O hydrogen bonding. In 4-nitroaniline, (I), each molecule is linked to four others by this means, and the overall supramolecular structure (Tonogaki et al., 1993) consists of sheets built from a single type of R44(22) ring (Bernstein et al., 1995). We have recently begun an exploration of the supramolecular structures of some analogues of 4-nitroaniline, including 2-amino-4-butylamino-6-methoxy-5-nitropyrimidine, (II) (Glidewell et al., 2003b), 2-amino-4,6-dimethoxy-5-nitropyrimidine, (III) (Glidewell et al., 2003a), and 2-amino-6-nitro-1,3-benzothiazole, (IV) (Glidewell et al., 2001), which form hydrogen-bonded supramolecular structures in one, two and three dimensions, respectively. Thus in (II), the molecules are linked by paired N—H···O hydrogen bonds to form a C(8) C(8)[R22(6)] chain of rings, while in (III), a combination of one N—H···N hydrogen bond and one N—H···O hydrogen bond generates a sheet built from alternating R22(8) and R66(32) rings. A three-dimensional framework is formed in (IV), built from a combination of one three-centred N—H···(O)2 hydrogen bond and one two-centred N—H···N hydrogen bond.
Continuing with this study, we have now investigated the molecular and supramolecular structures of 2-amino-5-nitrothiazole, (V), which crystallizes from ethanol solution as a monosolvate, C3H3N3O2S·C2H6O, (VI) (Fig. 1), in which the supramolecular structure takes the form of triply interwoven sheets.
The dimensions of the 2-amino-5-nitrothiazole component of (VI) show some unusual values, which are not consistent with the classically localized form (V) as the dominant contributor to the overall molecular-electronic structure. In particular, the two C—N bond lengths in the thiazole ring are effectively identical (Table 1), while the C—C bond is very long for a double bond of this type [mean value 1.326 Å, upper-quartile value 1.334 Å (Allen et al., 1987)]. Moreover, the C—NH2 bond is marginally shorter than the lower-quartile value (1.317 Å) for bonds of this type in enamine systems and significantly lower than the lower-quartile value (1.340 Å) for Caryl—NH2 bonds; the C—NO2 bond is very much shorter than typical Caryl—NO2 bonds (mean value 1.468 Å, lower-quartile value 1.460 Å), while the N—O bonds are both long for their type; finally, the C—S bonds are both much shorter than the lower-quartile value (1.809 Å) for single bonds between three-connected C and two-connected S atoms. Further insight into the molecular-electronic structure can be gained by using these bond lengths to estimate the corresponding bond orders using the recent recalibration by Kotelevskii & Prezhdo (2001) of the original equation relating bond order to bond length (Gordy, 1947). For the sequence of C—N and C—C bonds between amino atom N2 and nitro atom N1, the bond orders so calculated are, respectively, 1.83, 1.62, 1.61, 1.87 and 1.43, while the two C—S bond orders are 1.38 and 1.29. These data, taken together, indicate that the aromatic form (Va) and the polarized form (Vb) are both significant contributors to the molecular-electronic structure. The molecular dimensions in (IV) provide evidence for a similar type of polarization in that compound (Glidewell et al., 2001). Since the interbond angles at three-connected C atoms and at two-connected N atoms are optimally ca 120°, the constraints of a planar five-membered ring combined with the fact that the C—S bond distances are significantly longer than the other ring bonds leads to an interbond angle at S that is somewhat less than 90°, consistent with the use of only p orbitals by the S atom in the formation of the σ framework (Table 1). Consistent with the extensive conjugation, (Vb), the nitro group is effectively coplanar with the ring, as shown by the torsional angles (Table 2). The dimensions of the ethanol component are unexceptional.
The supramolecular structure of (VI) is complex, but it can readily be analysed by consideration, in turn, of each of the hydrogen bonds (Table 2). Amino atom N2 in the molecule at (x, y, z) acts as a hydrogen-bond donor, via atom H2A, to ring atom N3 in the molecule at (1 − x,1 − y,1 − z), so generating by inversion a centrosymmetric dimer, centred at (1/2,0.5, 1/2) and characterized by an R22(8) motif (Fig. 2). A similar motif also occurs in both (III) and (IV).
The same amino N2 atom at (x, y, z) also acts as a hydrogen-bond donor, this time via atom H2B, to ethanol atom O3, also at (x, y, z) (Fig. 1), and it is the ethanol molecules that link the R22(8) dimers into sheets. Ethanol atom O3 acts as a hydrogen-bond donor to the two nitro O atoms (O1 and O2) in the molecule at (x,-0.5 − y,-0.5 + z) in a planar, but very asymmetric, three-centred interaction (Table 2). The longer component of this system could, with some plausibility, be regarded merely as an adventitious contact. The thiazole molecule at (x,-0.5 − y,-0.5 + z) is a component of the R22(8) dimer centred at (1/2,-1,0), and propagation by the space group of this three-centred interaction links the dimer at (1/2,0.5, 1/2) directly to those centred at (1/2,-1,0), (1/2,-1,1), (1/2,2,0) and (1/2,2,1), thereby generating a deeply-puckered (100) sheet built from a combination of R21(4), R22(8) and R1010(38) rings, the latter two of which are both centrosymmetric (Fig. 3). The aforementioned dimers are themselves directly linked to the dimers centred at (1/2,0.5,-0.5), (1/2,0.5,1.5), (1/2,-2.5, 1/2) and (1/2,3.5, 1/2). Thus the repeat pattern of the sheet spans one unit cell in the [001] direction but three unit cells in the [010] direction (Fig. 4) and, accordingly, three such sheets are required to define the crystal structure fully. Since each sheet occupies the entire domain of x (namely −0.02 < x < 1.02), it follows that the sheets are threefold interwoven.
Within each set of three interwoven sheets, there are two weak interactions that link the sheets. Firstly, ring atom C3 in the thiazole molecule at (x, y, z), which is a component of the R22(8) dimer centred at (1/2,0.5, 1/2), acts as a hydrogen-bond donor to nitro atom O1 in the thiazole molecule at (1 − x,0.5 + y,1.5 − z), which forms part of the sheet containing an R22(8) dimer centred at (1/2,-0.5, 1/2). Secondly, the thiazole rings in the molecules at (x, y, z) and (-x,1 − y,-z), which lie in adjacent sheets of an interwoven triple, are parallel, with an interplanar spacing of 3.343 (3) Å and a centroid separation of 3.700 (3) Å, corresponding to a centroid offset of 1.586 (3) Å. However, although the sheets of an interwoven set are all weakly linked by these interactions, there are no direction-specific interactions between one triply interwoven set of sheets and the two adjacent sets, so that the supramolecular structure is strictly two-dimensional.
The supramolecular structure in (VI) thus stands in contrast to those in the simple solvent-free analogues (I)–(IV); in particular,the two dimensional aggregation in (VI) may be contrasted with the three-dimensional aggregation in the benzo analogue (IV). The aggregation in (VI) may also be contrasted with that observed in the 1:1 adduct, (VII), formed between (V) and 4-aminobenzoic acid [CSD (Allen, 2002) refcode MIRQEJ; Lynch (2001)]. Adduct (VII) crystallizes in the acentric space group P21, so that centrosymmetric hydrogen-bonding motifs cannot occur. The aggregation is dominated by the linking of the two neutral molecular components by means of N—H···O and O—H···N hydrogen bonds, and these two-component aggregates are then linked into spiral chains by a second N—H···O hydrogen bond.