

 journal menu
journal menu research papers
research papers









The earlier reported structure of the title compound, disodium tetracyanonickelate(II) trihydrate, Na2Ni(CN)4.3H2O, has been found to be incorrect and has now been redetermined. The charge distribution has been determined by multipole refinements against single-crystal X-ray diffraction data. In the refinement based on 30 K data a comparison was made between the results obtained using hydrogen positions and displacement parameters from X-ray diffraction with those using the values determined by neutron diffraction. The spin density was investigated by polarized neutron diffraction at 1.6 K. Crystal data: at T = 30 K: a = 7.278 (4), b = 8.856 (5), c = 15.131 (8) Å, α = 89.32 (5), β = 87.39 (4), γ = 83.61 (4)°; at 295 K: a = 7.392 (4), b = 8.895 (4), c = 15.115 (8) Å, α = 89.12 (2), β = 87.46 (2), γ = 84.54 (2)°. The structure contains practically square planar Ni(CN)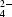 ions, which are stacked on top of each other in almost linear chains along the a direction. The separation between the Ni(CN) planes is rather large, with Ni—Ni distances around 3.7 Å. The six crystallographically independent water molecules are each coordinated to two sodium ions, approximately in the tetrahedral (lone-pair) directions, and the polarizing influence of these sodium ions also appears to be reflected in the deformation density in the lone-pair plane. The charge density based on the deformation functions of all atoms in the structure is compared with the individual densities calculated from the deformation functions of only nickel or the separate water molecules. In this way the effects of simple superposition of the individual densities have been studied. In the planar Ni(CN) ion the individual deformation density of nickel is in qualitative agreement with that expected from crystal-field theory. As the repulsion from the electrons is much weaker perpendicular to the Ni(CN) plane than within this plane, the deformation density is considerably larger in the perpendicular direction. However, the largest maxima in the individual deformation density around nickel are not found precisely in the planes defined by nickel and the four cyanide ligands or in the perpendicular direction just mentioned, which illustrates that it is necessary to consider the crystal field due to the whole crystalline environment.
ions, which are stacked on top of each other in almost linear chains along the a direction. The separation between the Ni(CN) planes is rather large, with Ni—Ni distances around 3.7 Å. The six crystallographically independent water molecules are each coordinated to two sodium ions, approximately in the tetrahedral (lone-pair) directions, and the polarizing influence of these sodium ions also appears to be reflected in the deformation density in the lone-pair plane. The charge density based on the deformation functions of all atoms in the structure is compared with the individual densities calculated from the deformation functions of only nickel or the separate water molecules. In this way the effects of simple superposition of the individual densities have been studied. In the planar Ni(CN) ion the individual deformation density of nickel is in qualitative agreement with that expected from crystal-field theory. As the repulsion from the electrons is much weaker perpendicular to the Ni(CN) plane than within this plane, the deformation density is considerably larger in the perpendicular direction. However, the largest maxima in the individual deformation density around nickel are not found precisely in the planes defined by nickel and the four cyanide ligands or in the perpendicular direction just mentioned, which illustrates that it is necessary to consider the crystal field due to the whole crystalline environment.





